
Abstract
Guadiamar River is located in the southwest of the Iberian Peninsula and connects two protected areas in the South of Spain: Sierra Morena and Doñana National Park. It is sited in an area affected by urban, industrial and agriculture sewage pollution and with tradition on intensive mining activities. Most of the studies performed in this area have been mainly focused on the presence of heavy metals and, until now, little is known about the occurrence of other contaminants such as emerging organic pollutants (EOPs). In this work, an analytical method has been optimized and validated for monitoring of forty-seven EOPs in surface water. The analytical method has been applied to study the distribution and environmental risk of these pollutants in Guadiamar River basin. The analytical method was based on solid-phase extraction and determination by liquid chromatography-triple quadrupole–tandem mass spectrometry. The 60 % of the target compounds were found in the analyzed samples. The highest concentrations were found for two plasticizers (bisphenol A and di(2-ethyhexyl)phthalate, mean concentration up to 930 ng/L) and two pharmaceutical compounds (caffeine (up to 623 ng/L) and salicylic acid (up to 318 ng/L)). This study allowed to evaluate the potential sources (industrial or urban) of the studied compounds and the spatial distribution of their concentrations along the river. Environmental risk assessment showed a major risk on the south of the river, mainly due to discharges of wastewater effluents.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Domestic and industrial activities are often the main sources of pollutants into the aquatic environment. Pesticides, polycyclic aromatic hydrocarbons and polychlorinated biphenyls are some examples of classic organic pollutants frequently detected and monitored in aquatic systems (Oliveira et al. 2011; Pitarch et al. 2016). However, the increasing production and usage of new products, as plastic and textile materials, fragrances, deodorants and other personal care products, non-stick cookware, etc. has resulted in continuous discharges of hundreds of new chemical pollutants to the aquatic environment. Some emerging organic pollutants (EOPs), for example, pharmaceutically active compounds (Camacho-Muñoz et al. 2010; Martín et al. 2011), personal care products (Terasaki et al. 2009), plasticizers (Staniszewska et al. 2016), brominated flame retardants (Vorkamp et al. 2014), perfluorinated compounds (Arvaniti and Stasinakis 2015) and surfactants (Corada-Fernández et al. 2011; Jurado et al. 2012) among others, have been found in wastewater (Arvaniti and Stasinakis 2015; Santos et al. 2009), surface water (SW) (Haman et al. 2015; Meffe and Bustamante 2014; Tijani et al. 2016) or sediments (Camacho-Muñoz et al. 2010) at concentrations from ng to hundreds of micrograms per litre.
Concern for some of these pollutants led to the European Union to increase the list of 33 priority pollutants included in the Decision 2455/2001/EC (EC 2001) to 45 compounds (Directive 2013/39/EU, EC 2013) and to set limit values for their concentrations in SW. Among the organic compounds added to the list of priority substances are biocides, perfluorinated compounds (PFCs) (perfluorooctane sulfonic acid (PFOS) and its derivatives) and brominated flame retardants (hexabromocyclododecane (HBCDD)). The first “watch list” of substances for European Union monitoring in the field of water policy was published in 2015 (EU Decision 2015/495) (EC 2015). This first "watch list" includes the pharmaceutical compound diclofenac and the hormones 17α-ethinylestradiol, 17β-estradiol and estrone.
However, despite the efforts of most of the international organizations in the field of water policy, such as the World Health Organization, the United States Environmental Protection Agency and the European Commission, many organic pollutants reaching the aquatic environment are not properly regulated. The main problems for the inclusion of these EOPs in the list of priority pollutants are the lack of information about their sources, occurrence, distribution and potential environmental risks and the lack of reliable analytical methodologies for their adequate determination.
The Guadiamar River is one of the major tributaries of the final stretch of Guadalquivir River (South of Spain). It connects two protected areas: Sierra Morena and Doñana National Park (declared a Biosphere Reserve and Human Heritage site by UNESCO in 1994).
Guadiamar River is affected by urban (in the lower reaches of the river), agriculture (mainly from its affluent streams Ardachón and Alcarayón) and industrial sewage pollution (in its North area). In fact, mining activities, sited in its North area, have traditionally been its main source of contamination. As a result, most of the pollution studies performed in Guadiamar River have mainly been focused on the presence of heavy metals, especially in soils and sediments. However, little is known about the concentrations and patterns of other contaminants such as EOPs.
The aims of this work were (i) to select a list of the most problematic EOPs in SW according to their persistence on the environment, toxicity and concentrations and frequency of detection in SW, (ii) to develop and validate an analytical method for the determination of the selected compounds, (iii) to obtain a comprehensive overview of the river water quality related to the presence of these pollutants through the study of their potential sources, their occurrence and spatial distribution in SW and their potential ecotoxicological risks.
Experimental
Selection of analytes
The selection of the target pollutants was carried out taking into account their persistence on the environment (Jurado et al. 2012; Meffe and Bustamante 2014; Tijani et al. 2016; Zareitalabad et al. 2013; Vorkamp et al. 2014), toxicity (Hernando et al. 2006; Terasaki et al., 2009; Yamamoto et al. 2011; Campo et al., 2015), concentrations and frequency of detection in SW (Arvaniti and Stasinakis, 2015; Camacho-Muñoz et al. 2010; Campo et al. 2015; Corada-Fernández et al. 2015; Haman et al. 2015; Jurado et al. 2012; Peng et al., 2008; Tijani et al. 2016; Vorkamp et al. 2014; Zolfaghari et al. 2014) as well as the recent legal requirements in the field of water policy (EC 2013; EC 2015). According to these criteria, several families of organic pollutants, with urban, rural and industrial use, were selected (Table 1). These compounds were classified as (i) pharmaceutically active compounds for human use (acetaminophen, diclofenac, ibuprofen, ketoprofen, naproxen, salicylic acid, sulfamethoxazole, trimethoprim, carbamazepine, atenolol, propranolol, caffeine, clofibric acid, bezafibrate, gemfibrozil), (ii) pharmaceutically active compounds for veterinary use (sulfadiazine, sulfamethazine, oxytetracycline, norfloxacin, ofloxacin, ciprofloxacin, tetracycline, doxycycline, chlortetracycline) and (iii) hormones (17α-ethinylestradiol, 17ß-estradiol, estriol, estrone) and industrial pollutants including the surfactants linear alkylbenzene sulfonates (LAS C10, LAS C11, LAS C12 and LAS C13), nonylphenol ethoxylates (NPE) (monoethoxylate (NP1EO), diethoxylate (NP2EO) and nonylphenol (NP)), mainly originated as degradation products of nonylphenol polyethoxylate surfactants), preservatives (methylparaben (MeP), ethylparaben (EtP) and propylparaben (PrP)), the PFCs (perfluorooctanesulfonic acid (PFOS), perfluorooctanoic acid (PFOA), perfluoroheptanoic acid (PFHpA), perfluorohexanoic acid (PFHxA), perfluoropentanoic acid (PFPeA) and perfluorobutanoic acid (PFBuA)) used as additives in various products such as cleaning products, fire-fighting foams, lubricants and paints, the plasticizers (bisphenol A (BPA) and di-(2-ethylhexyl)phthalate (DEHP)) and the flame retardant HBCDD.
Chemicals and reagents
The analytical standards (purity grade >97 %) and the internal standards bisphenol A d14, perfluorooctanoic acid 13C4, phenacetin-ethoxy-1-13C and propyl 4-hydroxybenzoate-13C6 were purchased from Sigma–Aldrich (Madrid, Spain) and Dr. Ehrenstorfer (Augsburg, Germany).
Three-millilitre solid-phase extraction (SPE) cartridges packed with 60 mg of Oasis HLB were purchased from Waters (Milford, MA, USA).
Individual stock standard solutions of each compound (1000 mg/L) and internal standard solutions (100 mg/L) were prepared in methanol and stored at 4°C. Working solutions of target compounds (10 mg/L) were prepared by dilution of stock standard solutions with methanol. Working solutions and the individual stock standard solutions of internal standards were used to obtain spiked samples.
Mixtures of the studied compounds at different concentration levels, containing 200 ng/L of each internal standard, were freshly prepared by appropriate dilutions of the stock standard solutions in methanol:water 50:50 (v/v). These solutions were used as calibration standards.
HPLC-grade acetonitrile, methanol and water were supplied by Romil Ltd. (Barcelona, Spain). Analytical grade hydrochloric acid (37%) and formic acid (98%) were obtained from Panreac (Barcelona, Spain). Ammonium formate and ammonium acetate were purchased from Sigma-Aldrich (Steinheim, Germany).
Studied area and sampling
The Guadiamar River (South of Spain) is one of the major tributaries of the final stretch of Guadalquivir River and, together with its two major tributaries (Agrio River and Alcarayon stream), constitutes one of the major water input to Doñana National Park. Regarding human activity, Guadiamar River basin (1319 m2) has traditionally been affected mainly by two pollution sources: mining (northern section) and urban-industrial and agricultural activities (southern section). It is affected by 22 municipalities whose main activity is agriculture and, in a lower extent, textile, food industry and mining.
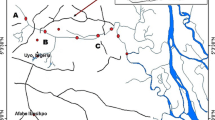
Water samples were collected in June 2014 from six different sampling sites located in the Guadiamar River and its tributaries (Figure 1). Sampling site S1 (37° 31’ 25.69” N, 6° 11’ 21.47” W) (nearby Gerena ≈6000 inhabitants) is located in the Guadiamar River, upstream of the influence of a pyritic mine (Aznalcóllar). Site S2 (37° 23' 46.36" N, 6° 13' 45.45" W) is situated after the confluence of the Agrio River with the Guadiamar River. This sampling point is affected, through Agrio River, by wastewater effluent discharges and a hazardous waste treatment plant sited in Aznalcollar. Site S3 (37° 18’ 10.44” N, 6° 15’ 35.32” W) is located after the confluence of Ardachón stream. These locations (S2 and S3) present the highest population density: Aznalcóllar (≈6000 inhabitants) and Sanlúcar la Mayor (≈12000 inhabitants) in S2 and Aznalcázar (≈4000 inhabitants) and Pilas (≈12000 inhabitants) in S3. Sites S4 (37° 10’ 52.41” N, 6° 11’ 54.88” W) (nearby Villamanrique de la Condesa ≈4000 inhabitants), S5 (37° 8’ 51.37” N, 6° 11’ 53.94” W) (nearby Isla Mayor ≈5800 inhabitants) and S6 (37° 6’ 21.85” N, 6° 15’ 30.20” W) are situated after the confluence of the Alcarayón streams and the Guadiamar River. Beside of wastewater effluent discharges and the industrial activities sited in the north area, Guadiamar River is affected by diffuse pollution sources from agricultural activities sited in the South of the river (sampling points S3 to S6).

Sampling sites along the course of Guadiamar River. (WWTP: wastewater treatment plant; HWTP: hazardous waste treatment plant)
Each composite sample (2.5 L) was collected by mixing five grab samples collected across the river in each sampling point. These samples were collected one after the other in a short period of time. Samples were transported to the laboratory in 2.5 L amber glass bottles, pre-cleaned with acetone and methanol, pre-rinsed with SW from the site sampled and refrigerated (4°C). Samples were stored at -30°C until analysis (within 48 h after sample collection).
Sample treatment
Optimization of sample treatment was carried out evaluating the influence of sample pH (pH 2 and 7) and elution solvent (acetone and methanol). Extraction of the studied compounds was carried out using Oasis HLB SPE cartridges, previously conditioned with successive aliquots of 3 mL of methanol and 3 mL of water (acidified to pH 2 with hydrochloric acid). 250 mL of SW samples were filtered through a 1.2 μm glass fibre membrane filter (Whatman, Mainstone, UK) and acidified to pH 2 with hydrochloric acid. Then, the internal standards were added to achieve individual concentrations of 0.4 ng/L. Samples were percolated through the cartridges at a flow rate of about 10 mL/min using a vacuum manifold system (Waters, USA) connected to a vacuum pump. Cartridges were rinsed using 3 mL of methanol:water (5:95 v/v) acidified to pH 2. Elution of the studied compounds was performed with three successive aliquots of 1 mL of methanol at a flow-rate of about 1 mL/min. The combined aliquots were evaporated to dryness by a gentle nitrogen stream and the residue was dissolved in 0.3 ml of methanol:water (50:50, v/v), filtered through a 0.22 μm nylon filter and immediately analyzed by liquid chromatography-triple quadrupole mass spectrometry (LC-QqQ-MS/MS).
LC-QqQ-MS/MS determination
After sample treatment, three aliquots of 20 μL of each sample extract were analyzed by LC-QqQ-MS/MS. Three chromatographic methods were used, one method for the determination of (i) pharmaceuticals for human use, (ii) a second method for the determination of veterinary pharmaceuticals and (iii) a third method for the determination of the hormones and industrial pollutants, including BPA, DEHP, HBCDD, NPE, LAS, PFCs and parabens. Target compounds were classified into three groups because of their different LC-MS/MS behaviour. Each analysis was carried out in triplicate to avoid false positives.
Chromatographic separations were performed on an Agilent 1200 series HPLC system (Agilent, USA) equipped with a vacuum degasser, a binary pump, an autosampler and a thermostated column compartment.
MS analyses were done with a 6410 QqQ instrument equipped with an electrospray ionization source (ESI) (Agilent). Ionization of analytes was carried out using the following settings: MS capillary voltage 3000 V, drying-gas flow rate 9 L/min, drying-gas temperature 350 °C and nebulizer pressure 40 psi. Detection was performed in multiple reaction-monitoring mode (MRM). MS/MS parameters were optimized by the injection without column of individual stock standard solutions of target compounds and internal standards (10 mg/L) using different combinations of aqueous phase (water (0.1 % formic acid), aqueous solution of ammonium acetate, aqueous solution of sodium formate (0.1 % formic acid)) and organic phase (methanol or acetonitrile (0.1 % formic acid)). Considering their abundances, two transitions were selected for each compound. The most intensive transition was used for the quantification and the less intense transition and the relation between both transitions were used for the confirmation of the analytes. Table 2 shows the optimized MS/MS parameters for MRM analysis and the internal standards used in each method. Chromatographic separations were optimized using different gradients with the selected mobile phases for each group of compounds. Retention times of target compounds and chromatograms of the standard solutions are shown in supplementary materials (Tables S1 and S2 and Figures S3-S5).
Quantification of the target compounds was carried out using the internal standard method. Calibration curves were constructed by linear regression of the peak area ratio of the analyte and its corresponding internal standard against their respective concentrations. Calibration curves were constructed in the concentration range expected for each compound in SW.
Analysis of human pharmaceuticals
Separation of pharmaceutically active compounds for human use was carried out using a HALO C18 (50x4.6 mm i.d.; 2.7 μm) analytical column (Teknokroma, Spain) protected by a HALO C18 (5x4.6 mm i.d.; 2.7 μm) guard column (Teknokroma, Spain). Analytes were separated by gradient elution with (A) acetonitrile (containing 0.1% v/v formic acid) and (B) aqueous 10 mM ammonium formate solution (containing 0.1% v/v formic acid) at a flow rate of 0.6 mL/min. Column temperature was kept at 30 °C. Elution program was as follow: 0-2 min isocratic 10 % of solvent A; 2-13 min, linear gradient from 10 to 90 % of solvent A; 13-15.5 min isocratic conditions at 90 % of solvent A and, finally, back to 10% of organic phase in 2 min.
Analysis of veterinary pharmaceuticals
Separation of veterinary pharmaceuticals was carried out using a Zorbax Eclipse XDB–C18 Rapid Resolution HT (50x4.6 mm i.d.; 1.8 μm) analytical column (Agilent, USA). Analytes were separated by gradient elution with (A) acetonitrile (containing 0.1% v/v formic acid) and (B) water (containing 0.1% v/v formic acid) at a flow rate of 0.6 mL/min. Column temperature was kept at 30 °C. Elution program was as follow: 0-10 min isocratic 10 % of solvent A; 10-13 min linear gradient from 10 to 30 % of solvent A; 13-18 min isocratic conditions at 30 % of solvent A and, finally, back to 10 % of solvent A in 2 min.
Analysis of industrial EOPs
Industrial EOPs were separated using a HALO C18 (50x4.6 mm i.d.; 2.7 μm) analytical column protected by a HALO C18 (5x4.6 mm 1.d.; 2.7 μm) guard column. Separation was carried out by gradient elution with (A) methanol and (B) aqueous 10 mM ammonium acetate solution at a flow rate of 0.6 mL/min. Column temperature was kept at 30 °C. Elution program was as follow: 0-14 min linear gradient from 28 to 70 % of solvent A; 14-19 min linear gradient from 70 to 80 % of solvent A; 19-25 min linear gradient form 80 to 100 % of solvent A; 25-27 min isocratic conditions at 100 % of solvent A, and, finally, back to 28 % of organic phase in 2 min.
Method validation
Analytical method was validated by the determination of extraction process recovery, matrix effect, process efficiency, precision (expressed as repeatability in terms of relative standard deviation (RSD)), linearity and method detection limit (MDL) and method quantification limit (MQL).
Extraction process recovery was evaluated by comparison of the peak areas of the analyte in samples spiked before and after extraction (solutions A and B, respectively): R (%) = (A/B) x 100. Blank samples (non-spiked samples) were measured to apply blank corrections to signals obtained from spiked samples.
Matrix effect was calculated as previously described by Martín et al. (2014). Matrix effect was determined by comparison of the peak areas of samples spiked after extraction (B), peak areas of blank samples (non-spiked sample) (C) and peak areas from a standard solution at the same concentration level (D). The matrix effect (ME) is expressed as a percentage as follows: ME (%) = (B-C)/D x 100. If ME is higher than 100, signal enhancement due to matrix effect has occurred. If ME is lower than 100, signal suppression due to matrix effect has occurred.
Process efficiency were calculated as reported previously by (Matuszewski et al. 2003) considering matrix effect and the recovery of the extraction process: PE (%) = (ME x R)/100.
Precision of the method was calculated as the RSD of the concentrations determined in a spiked sample measured in triplicate. Linearity of the method was determined by analysing standard solutions in triplicate at different concentration levels in the range of concentrations expected after extraction. The internal standard method was used for the quantification of the target compounds, except for LAS and DEHP, in sample extracts because it was demonstrated to be the most suitable calibration approach for their quantification. Calibration curves were constructed by linear regression of the peak area ratio of the analyte and its assigned internal standard against their respective concentrations. No suitable matrix effect correction was obtained for LAS and DEHP when using internal standard calibration, therefore, external calibration method was applied. Matrix effect correction of LAS and DEHP signals was carried out taking into account matrix effect values obtained in method validation. The concentrations of the target compounds in the studied samples were calculated from the concentrations determined in sample extracts taking into account the recoveries and matrix effects. Instrumental detection (IDL) and quantification (IQL) limits were estimated as the concentrations of each analyte corresponding to a signal-to-noise ratio of 3:1 and 10:1, respectively. IDL and IQL were determined by the injection of sample extracts spiked in triplicate at low concentration levels (from 0.01 to 250 μg/L) after solid-phase extraction. In the case of compounds, such as DEHP, that were present in blank extracts, IDL and IQL were estimated as the concentrations corresponding to blank signal plus three times or ten times the standard deviation of blank signal, respectively. Method detection (MDL) and quantification limits (MQL) were determined applying the enrichment factor and recoveries achieved in solid-phase extraction to the IDL and IQL.
Quality Control
In each batch of samples, one sample was divided into four subsamples. Three of them were spiked with the studied compounds and matrix effect and recoveries were determined and compared with those achieved during validation process.
Each batch contained calibration standards, spiked samples, samples, procedural blank and standard quality control. Procedural blank and standard quality control were measured before and after the calibration standards and every five samples.
Ecotoxicological Risk Assessment
Environmental risk assessment was carried out using risk quotient (RQ) values according to the European Union Technical Guidance Document (EC-TGD, 2003; Tauxe-Wuersch et al., 2005; Gros et al., 2010; Mendoza et al., 2015). RQs were calculated for each compound as the ratio of the measured environmental concentration (MEC) and the predicted no effect concentration (PNEC).
Concentrations measured during the monitoring period were used as MEC values. PNEC values were calculated, according to the European Union Technical Guidance Document, dividing the lowest ecotoxicological data, of acute and chronic toxicity studies available in the literature for several species representing different trophic levels (normally algae, bacteria, invertebrate and fish species), by an assessment factor (AF). No observed effect concentration (NOEC) was used for chronic toxicity. For acute toxicity, the effect concentration (EC50), that refers to the concentration at which the 50% of its maximal effect was observed in test species after a specified test duration or the lethal concentration (LC50), that refers to the concentration that causes the death of the 50 % of a group of test species after a specified test duration, were applied.
An AF of 1000 was applied when at least one short-term L(E)C50 from each of the three evaluated trophic levels was available. An AF of 100 was applied when one long-term NOEC value was available for algae, crustaceans or fish. An AF of 50 was applied when two long-term NOEC values were available for species in two different trophic levels. An AF of 10 was used when NOEC values for species in the three evaluated trophic levels were available (EC 2003).
An exhaustive literature review was performed to collect toxicity data of the studied compounds in species at different trophic levels. Toxicity data applied and PNEC values of detected compounds are presented in Tables S3 and S4, respectively, in supplementary data.
RQ values higher than one imply significant ecotoxicological risk to aquatic organisms. Common criteria to interpret RQ values, in risk assessment studies, establish different risk levels: low risk (RQ values from 0.01 to 0.1), medium risk (RQ values between 0.1 and 1) and high risk (RQ values higher than 1) (Hernando et al. 2006).
Results and discussion
Optimization of solid-phase extraction
Optimization of solid-phase extraction was carried out using Milli-Q water spiked with the target compounds at 500 ng/L. Oasis HLB cartridges were selected because of their suitability to extract acid, neutral and basic compounds. Conditioning, washing procedure, and flow rates applied were those recommended by the manufacturer. Sample pH (pH 2 and 7) and elution solvent (acetone and methanol) were tested. Sample pH was optimized using six aliquots of 250 mL of Milli-Q water spiked with the target compounds. Sample pH was adjusted to 2 in three of the spiked aliquots using 3 M hydrochloric acid solution. Samples were percolated through the cartridges at a flow rate of about 10 mL/min using a vacuum manifold system (Waters, USA) connected to a vacuum pump. Cartridges were rinsed using 3 mL of methanol:water (5:95 v/v). Elution of the target compounds was performed with three successive aliquots of 1 mL of methanol at a flow-rate of about 1 mL/min. The combined aliquots were evaporated to dryness by a gentle nitrogen stream and the residue was dissolved in 0.3 mL of methanol:water (50:50, v/v), filtered through a 0.22 μm nylon filter and analysed by liquid chromatography-triple quadrupole mass spectrometry (LC-QqQ-MS/MS).
Figure S1 (supplementary material) shows the recoveries achieved at the tested sample pH values. The highest recoveries were achieved for acidified samples, especially in the case of pharmaceuticals ketoprofen, naproxen, sulfamethoxazol, caffeine and salicylic acid and LAS C13 and PFBuA. Because of that, acidification of samples to pH 2 was selected.
Figure S2 shows the recoveries achieved using acetone and methanol as elution solvents. For most of the studied compounds, the recoveries achieved using methanol as elution solvent were slightly higher than those obtained using acetone especially in the case of PFBuA, PFPeA (Figure S2). According to these results, acidification of samples to pH 2 and the use of methanol as extraction solvent were selected for the SPE procedure (Figure S2).
Method performance
Table 3 shows process efficiency (%), recovery (%), matrix effect, precision, linearity and limits of detection and quantification of the proposed analytical method.
Process efficiencies achieved in the optimized analytical method were in the range from 62 (chlortetracycline) to 112 % (norfloxacyn). Only acetaminophen, atenolol and PFBuA were poorly recovered (Table 3). In the case of acetaminophen and atenolol, the low recoveries achieved (31 and 24 %, respectively) could be due to the sample pH applied to the solid-phase extraction. Other authors have reported higher recoveries of these compounds using higher pH values (Grujić et al. 2009; Santos et al. 2013; Vieno et al., 2006). The low recovery of PFBuA can be related to its higher hydrophilicity that can results in its elution during the wash of the SPE cartridge (Martín et al. 2014).
Matrix effect was evaluated by comparison of the signals obtained in samples spiked after solid-phase extraction with those obtained in standard solution at the same concentration. For most of the studied compounds, signal suppression was observed. Only in the case of salicylic acid, caffeine, 17α-ethynylestradiol, clofibric acid, DEHP and MeP, signal enhancement was observed. The highest signal suppression was observed for 17β-estradiol (77 %) and estriol (66 %). The highest signal enhancement was observed for DEHP (139 %). Matrix effect for the other target compounds was in the range 80-119 %. Therefore, matrix-matched calibrations were considered no necessary. Concentration of each compound in the SW samples was determined from the concentration measured in the sample extract and taking into account matrix effect and recovery achieved after solid-phase extraction.
Instrumental inter- and intra-day precision were determined by the injection of standard solutions in methanol:water prepared in triplicate. Results are shown in the supplementary material (Tables S1 and S2). Precision of the method, measured as RSD, was lower than 25 %. Linearity, evaluated by the injection of nine calibration points for all compounds at concentrations from IQL to 2000 μg/L resulted in r2 >0.994 (Table 3).
MDL and MQL ranged from 0.02 to 98 ng/L and from 0.05 to 297 ng/L, respectively. In the case of DEHP (MQL 9.43 ng/L), HBCDD (0.80 ng/L), NP (27.0 ng/L) and PFOS (0.12 ng/L), the MQL values achieved allowed the determination of these compounds at the concentration limits fixed by the European Union in SW (1.3, 0.5, 2.0 and 36 μg/L in inland SW, and 1.3, 0.05, 2.0 and 7.2 μg/L in other SW, for DEHP, HBCDD, NP and PFC, respectively) (EC 2013). Moreover, MDL achieved for diclofenac (0.04 ng/L) complies with the requirements established by the EU Decision 2015/495 (EC 2015) (maximum acceptable method detection limit for diclofenac: 10 ng/L). MDL achieved for the hormones 17α-ethinylestradiol (15.0 ng/L), 17β-estradiol (2.13 ng/L) and estrone (0.62 ng/L) were higher than maximum acceptable method detection limits fixed by the European Decision 2015/495 (EC 2015) (17α-ethinylestradiol (0.035 ng/L), 17β-estradiol (0.4 ng/L) and estrone (0.4 ng/L)). This fact is due to the low ionization and fragmentation of these compounds under LC-MS/MS conditions applied for their determination (IDL: 12.8, 1.89 and 0.64 μg/L for 17α-ethinylestradiol, 17β-estradiol and estrone, respectively).
Concentrations of EOPs in Guadiamar River
Concentrations of target compounds found in studied samples are shown in Table S5 (supplementary material). Figure 2 shows the concentrations of the studied compounds in Guadiamar River basin as a box and whisker plot representing the 5 and 95 percentile. Only compounds with a frequency of detection higher than 30% were included in the figure.

Concentrations of LAS, NPE, plasticizers, pharmaceutically active compounds, PFCs and parabens in Guadiamar River basin
The higher concentrations were found for BPA, DEHP, NPE and LAS (mean concentrations from 133 to 930 ng/L) and for the pharmaceutical compounds caffeine and salicylic acid with mean concentrations of 623 and 318 ng/L, respectively. Other pharmaceuticals (trimethoprim, propranolol, carbamazepine and diclofenac), parabens and PFCs showed concentrations up to 49.6 ng/L (PFHpA). Approximately, the 45 % of the studied compounds (acetaminophen, atenolol, ibuprofen, ketoprofen, naproxen, sulfamethoxazole, clofibric acid, gemfibrozil, 17α-ethinylestradiol, 17ß-estradiol, estriol, estrone, sulfadiazine, oxytetracycline, norfloxacin, ciprofloxacin, tetracycline, doxycycline and chlortetracycline, PFBuA and HBCDD) were not detected in the studied area.
BPA and DEHP were found in all the analyzed samples. This fact could be explained by their wide use in industrial production and daily use products (bottles, digital media, toys, paints, construction materials, personal care articles and cosmetics (Domínguez-Morueco et al. 2014; Huang et al. 2012)) and their poor removal in wastewater treatment plants (Guerra et al. 2015; Tran et al. 2015; Zolfaguari et al. 2014). BPA concentrations up to 1661 ng/L (mean 490 ng/L) were found; however, most of them (70 %) were lower than 440 ng/L. These concentrations were slightly higher than those previously reported (100 to 250 ng/L) in SW close to the studied area (Ballesteros-Gómez et al. 2007). This fact could be explained by the proximity of wastewater treatment plants as it was described in Germany where concentrations measured (1927 ng/L) were close to those found in this work (Quednow and Püttmann 2008). Regarding to DEHP, included as a priority pollutant in the WFD in 2008, measured concentrations (between 390 and 1191 ng/L) were lower than environmental quality standards (EQS), fixed on 1300 ng/L. However, 30 % of these concentrations were close to this value (higher than 1000 ng/L), which could indicate the need for further attention to this compound.
NPEs were found in all analyzed samples at concentration ranges of 160-285, 85-202 and 118-2260 ng/L for NP1EO, NP2EO and NP, respectively (mean concentrations 133, 208 and 930 ng/L, respectively). The distribution found (NP>NP2EO>NP1EO) could be explained by the degradation of nonylphenol polyethoxylates, into NP2EO, NP1EO and, finally, into NP (Corada-Fernández et al. 2011). The concentrations found in this work were slightly higher than those found in other European countries such as France (Cailleaud et al. 2007), Italy (Vitaly et al. 2004), Switzerland (Jonkers et al. 2009) and Greece (Arditsoglou and Voutsa 2010). This fact could indicate the influence of wastewater discharges and other potential sources, such as their use in pesticide formulations. Alkylphenols can also be present in the atmosphere due to industrial processes, waste incineration, wastewater treatment processes, among other sources. Then, they can pollute SW by atmospheric deposition or rainfall events (Bergé et al. 2012). For example, Bressy et al. (2011) described that NP fluxes up to 84.0 μg/m2/year for atmospheric deposition and up to 190 μg/m2/year for stormwater in a small urban catchment near to Paris.
Considering the EQS fixed by the Annex X of the Directive 2013/39/CE (EC 2013), the limit value fixed as maximum concentration in SW (2000 ng/L in the case of NP) was exceeded only in one of the sampling points. However, the 80 % of the analyzed samples presented concentrations of NP higher than the EQS fixed as annual mean concentration.
LAS homologues were quantified in all analyzed samples at mean concentrations of 214, 320, 338 and 251 ng/L for LAS C10, C11, C12 and C13, respectively, and at maximum concentrations up to 491 ng/L (LAS C13). The distribution of these compounds measured in this work (mean 19, 28, 30 and 23 % for LAS C10, C11, C12 and C13, respectively) was close to those used in products such as household and laundry detergents or personal care products such as shampoos and shower gel. These results could reflect that effluents from WWTPs constitute the main source of these compounds to the studied area. In spite of that, the concentrations found in this work were significantly lower than those reported previously in water samples from other aquatic systems in Spain and Europe, where values typically ranged from low μg/L to higher than 1000 μg/L have been reported (Corada-Fernández et al. 2015). For example, concentrations up to 2677 μg/L have been reported in SW from Guadalete River (SW, Spain) which it is affected by the dischages of a WWTP serving a population of more than 200,000 inhabitants (Corada-Fernandez et al. 2011). The lower concentrations measured in this work (from 819 to 1615 ng/L) could be due to the low population around the studied area.
Among the 28 human and veterinary pharmaceuticals analyzed in these work, only five of them were detected in all analyzed samples (diclofenac, salicylic acid, carbamazepine, propranolol and caffeine) and four (trimethoprim, bezafibrate, sulfamethazine and ofloxacin) were present in less than the 30 % of the samples (mean concentrations 25.3, 139, 396 and 107 ng/L, respectively). Concentrations of the other studied pharmaceutical compounds were lower than detection limits in all analyzed samples.
Caffeine and salicylic acid were found at the highest concentrations, up to 2050 and 709 ng/L, respectively, while diclofenac, carbamazepine and propranolol were found at concentrations up to 27.3, 28.0 and 8.32 ng/L. This distribution could be related to the high consumption of these compounds, in coffee, tea or chocolate in the case of caffeine and in several domestic products, including cosmetics, personal care products and food preservatives in the case of salicylic acid. In the case of veterinary pharmaceuticals, only sulfamethazine and ofloxacin were detected (sampling points S3-S5). This fact could be related to its use as a veterinary pharmaceutical in poultry production units in this catchment area. The concentrations of the pharmaceutical compound diclofenac, included in the “watch list” published by the European Commission (EC, 2015), were higher than the limit of detection purposed by the Decision (10 ng/L) in all analyzed samples.
PFCs were detected in all analyzed samples at concentrations from 24.3 to 56.7 ng/L (sum of the six studied compounds), which show their wide use and persistence in environmental samples. The highest mean concentrations were found for PFHpA (16.9 ng/L), followed by PFOA (9.00 ng/L), PFHpA (7.90 ng/L), PFHxA (6.4 ng/L) and PFOS (2.9 ng/L). The concentrations of PFOS measured in this work were higher than the EQS, fixed by Directive 2013/39/EC (EC, 2013) as annual average concentration (0.65 ng/L), in the 67 % of the analyzed samples. However, these concentrations were lower than the EQS fixed as maximum allowable concentration (36000 ng/L) in this directive.
MeP and PrP were the predominant parabens in the studied SW (found in all analyzed samples at mean concentrations of 21.5 ng/L and 8.06 ng/L, respectively). EtP was found in the 50 % of the studied samples at concentrations lower than 2.67 ng/L. These concentrations were similar to those reported in countries such as Switzerland (Jonkers et al. 2009), Portugal (Jonkers et al. 2010) or USA (Renz et al. 2013); however, highest concentrations were found in Japan and China (Peng et al. 2008; Yamamoto et al. 2011). These concentrations were found in high densely populated areas with SW highly contaminated by treated or untreated domestic sewage with no sewerage system.
The distribution found in this work, in concordance with that reported previously (MeP>PrP>EtP), is close to the composition of paraben mixtures in common consumer products such as food and cosmetics (Haman et al. 2015). As a result, the distribution found in analyzed SW samples could indicate that wastewater effluent discharges are the main source of these pollutants to the SW (Haman et al., 2015).
Spatial distribution of EOPs along Guadiamar River
Different trends were observed for the studied compounds in the studied river (Figure 3). These distributions could be explained considering the different pollution sources affecting the studied area: North area (S1 to S2), traditionally affected by mining and industrial activities and south area (S3-S6) affected by the discharges of wastewater effluents from nearby population to the Guadiamar River (≈19000 inhabitans) or through any of their tributaries such as Molinillo stream (≈600 inhabitants), Alcarayon stream (≈18600 inhabitants) or Cigüeña stream (≈10000 inhabitants).

Concentrations of human pharmaceuticals, LAS (sum of LAS C10, C11, C12 and C13), parabens (sum of MeP, EtP and PrP), NPE (sum of NP2EO, NP1EO and NP), PFCs (sum of PFBuA, PFPeA, PFHxA, PFHpA, PFOS and PFOA), BPA and DEHP along Guadiamar River
The concentrations of LAS homologues, as well as those of parabens, increased from S1 to S5. This distribution could be explained considering the urban discharge as the main source of these contaminants and the higher population affecting the downstream studied area. However, concentrations of parabens measured in sampling point S6 were lower than those measured in S5. The decrease of concentrations measured in sampling point S6, could be due to their photodegradation (An et al. 2014; Zúñiga-Benitez and Poñuela 2016) or biodegradation (Haman et al. 2015) under aerobic conditions, as well as the dilution effect caused by the high flow of Guadiamar River in this point.
The concentrations of BPA and DEHP, decreased from S1 to S6, which could be explained by the higher contribution of industrial effluents (hazardous waste treatment plant, mining, foam and plastic industries), upstream sited, to the load of these compounds to the studied river. Concentrations of NPE and PFC, showed a similar behaviour than concentrations of DEHP, except for sampling point S3 (NP) and S5 (PFC), where concentrations were up to 2060 ng/L (NP) and 53.5 ng/L (ΣPFC). These higher concentrations could be related with the discharges of the industrial activities located in the towns nearby to these sampling points (Aznalcázar and Sanlucar la Mayor, in the case of S3, and Isla Mayor, in the case of S5). This fact has been pointed out in several works (Bergé et al. 2012; Camacho-Muñoz et al. 2014; Castiglioni et al. 2015) in which concentrations of these compounds reported in industrial wastewater effluents were higher than those in domestic wastewater effluents. For example, Berge et al. (2012) reported concentrations of NP in industrial wastewater effluents (up to 400 μg/L) higher than those measured in domestic wastewater effluents (up to 170 μg/L) and Cestiglioni et al. (2015) reported concentrations of PFOA about 50 times higher than those measured in domestic wastewater effluents.
The concentrations of the studied pharmaceutically active compounds did not show a clear behaviour along the studied area. Their concentrations were similar in all the analyzed samples, except for the pharmaceuticals diclofenac, propranolol and salicylic acid in sampling point 4; caffeine in sampling point S6; and carbamazepine concentrations that increased from S3 to S6. These results could be explained by the proximity of the discharge of wastewater effluents to sampling point S4. The concentrations measured in sampling points S5 and S6, were lower than those measured in S4. This fact could be due to attenuation processes such as biodegradation or retention in the sediments (da Silva et al., 2011).
Regarding to veterinary pharmaceuticals, only sulfamethazine was present at concentrations higher than the detection limit in sampling points S3 (365 ng/L) and S5 (427 ng/L) while only ofloxacin was detected in sampling point S4 (107 ng/L). This fact could be due to their use in poultry farms located in the sampling area and to the natural attenuation of the concentrations of the other sulphonamides, tetracycline or fluoroquinolones through mechanism such as sorption to suspended matter (da Silva et al., 2011), colloids (Yang et al., 2011) or sediments (da Silva et al., 2011; Zhou et al., 2011).
Environmental risk assessment
European guidelines (EC, 2003; EMEA, 2006) recommend the use of chronic toxicity to calculate PNEC values. However, due to the current limited availability of chronic toxicity data, short-term (EC50 and LC50) values are widely used to calculate PNEC values and to estimate potential adverse effects to aquatic organisms (Gros et al., 2010).
In this study, RQs were evaluated considering data from acute and chronic toxicity studies, the most sensitive organism and the maximum concentration levels detected for each compound, which represents the “worst-case scenario” (Mendoza et al., 2015). Toxicity data applied and PNEC values of detected compounds are presented in supplementary material (Tables S3 and S4). RQ values, calculated as described in Section 2.8., are shown in Figure 4. RQ values of caffeine and some well-known endocrine disruptor compounds (NPEs, BPA, DEHP, MeP and LAS C13) were higher than 1 when acute toxicity data was considered. When chronic toxicity data is considered, diclofenac, propranolol, bezafibrate, ofloxacin, NP, NP1EO, DEHP and BPA are the ones with a higher negative impact to Guadiamar River.

Risk quotients (RQ) calculated for acute and chronic toxicity of compounds detected in Guadiamar River
The specific toxicity of each compound for each single species makes that highly used compounds, such as LAS C10-C12, not always are related to a high environmental risk. Moreover, compounds detected at low concentrations, as diclofenac or propranolol, could imply a higher risk due to their higher toxicity. RQ values obtained for pharmaceutical compounds are consistent with other RQs reported in literature in surface water (Paiga et al., 2016; Kuzmanović et al., 2015; Moreno González et al., 2014; Vázquez-Roig et al., 2012; Ginebreda et al., 2010; Wang et al., 2010). In a recent study carried out in Spain, RQ values higher than 10 were obtained for the above mentioned pharmaceutical compounds plus acetaminophen, ibuprofen, naproxen, clarithromycin, ofloxacin, and trimethoprim in hospital effluents (Mendoza et al., 2015). Wang et al. (Wang et al., 2010) reported RQ values higher than one for diclofenac in two Chinese rivers whereas for the other detected pharmaceuticals were below 1. In a recent study carried out for 55 pharmaceuticals and personal care products in a municipal wastewater treatment plant in Greece, diclofenac was identified as the compound with the highest potential environmental risk (Papageorgiou et al., 2016).
RQs were also evaluated for every single aquatic species (Figure S6 in the supplementary material). Besides, site-specific concentrations of detected pollutants along the catchment area were considered. Results showed that RQs are highly dependent on the specie used for the assessment (Figure S6 in Supplementary material). In case of diclofenac, propranolol, LAS C13, NPE, MeP and BPA adverse effects were observed when specific species are considered. Among the eighteen species considered for risk assessment of diclofenac, only the fish Salmo trutta f. fario showed RQ > 1 (between 3.7 and 5.4) while for the others, mainly bacteria, algae and the fish (D. rerio), RQ values lower than 0.008 were obtained. A similar scenario was observed for propranolol (RQ > 1 was obtained for the fish O. latipes whereas RQ were lower than 0.008 for the other species), ofloxacin (RQ > 1 for the algae P. subcapitata) and MeP (RQ > 1 for the fish O. latipes). In the case of the industrial pollutants, the most affected species were the invertebrate M. bahía, affected by NPE, and the algae S. capricornutum affected by DEHP. BPA showed high risk for certain invertebrates and fish species were used. Nevertheless, the results did not allow establishing the most sensitive trophic levels.
It is noteworthy that the aquatic risk assessment, on the basis of RQ estimation, pointed out a highly possible risk in all locations where NP was detected, even if the annual average concentration established by Directive 2013/39/EC (0.3 μg/L) was not overcame (location S6: 118 ng/L, RQ=5.7). Similar results were obtained by Kuzmanović et al. (2015) for 200 organic micropollutants monitored in four rivers located in the Mediterranean side of the Iberian Peninsula. Together with pesticides, NP and octylphenol were identified as the most problematics for the studied rivers. In this paper, environmental risk assessment has been carried out considering the concentrations of EOPs during a limited period of time. A larger study, considering possible seasonal influences, would be necessary for a better understanding of the environmental impact of these compounds.
Moreover, RQs act as a normalized measurement of risk, thereby allowing a comparison between different compounds with different toxicities and exposure levels. However, since the scenario studied is characterised by the simultaneous presence of multiple compounds rather than a single product, estimation of the toxicity caused by pollutant mixtures is essential (Mendoza et al., 2015). Therefore, further studies are required for a better understanding of the environmental impact due to these compounds.
Conclusions
In this paper, an analytical method based on solid-phase extraction and determination by high-performance liquid chromatography with triple-quadrupole mass spectrometry detector has been applied to determine 47 organic EOPs in Guadiamar river basin (southwest, Spain). The spatial distribution of these compounds and their potential environmental risks have been evaluated.
Up to 60 % of the studied compounds were found in the analyzed samples. Plasticizers, surfactants and the pharmaceutically active compounds caffeine and salicylic acid were the compounds at the highest concentrations. Among the compounds included in the priority contaminant list, NP and PFOS were the ones at concentrations higher than the environmental quality standards, while other compounds, such as DEHP, were found at concentrations close to their environmental quality standards.
The distribution of the studied compounds in Guadiamar River showed both, industrial (on the north area of the river) and domestic discharges (on the south area). In the upper part of the river and in industrialized areas the presence of regulated priority pollutants were more predominant, whereas the lower part of the river was more affected by the presence of emerging and non-regulated priority pollutants due to a higher number of effluent wastewater discharges.
Regarding risk assessment, the calculated RQs were highly dependent on the specie used for the assessment. Considering the worst-case scenario, acute risk could be expected due to caffeine, NPEs, BPA, DEHP, MeP and LAS C13. Diclofenac, propranolol, bezafibrate, ofloxacin, NP, NP1EO, DEHP and BPA presented the highest negative impact to Guadiamar River when chronic toxicity data is considered. Moreover, NP could represent a high risk in all locations where it was detected, in spite of the annual average concentration established by Directive 2013/39/EC was not overcame.
References
An T, Fang H, Li G, Wang S, Yao S (2014) Experimental and theoretical insights into photochemical transformation kinetics and mechanisms of aqueous propylparaben and risk assessment of its degradation products. Environ Toxicol Chem 33:1809–1816
Arditsoglou A, Voutsa D (2010) Partitioning of endocrine disrupting compounds in inland waters and wastewaters discharged into the coastal area of Thessaloniki, Northern Greece. Environ Sci Pollut Res 17:529–538
Arvaniti OS, Stasinakis AS (2015) Review on the occurrence, fate and removal of perfluorinated compounds during wastewater treatment. Sci Total Environ 524–525:81–92
Ballesteros-Gómez A, Ruiz FJ, Rubio S, Pérez-Bendito D (2007) Determination of bisphenols A and F and their diglycidyl ethers in wastewater and river water by coacervative extraction and liquid chromatography–fluorimetry. Anal Chim Acta 603:51–59
Bergé A, Cladière M, Gasperi J, Coursimault A, Tassin B, Moilleron R (2012) Meta-analysis of environmental contamination by alkylphenols. Environ Sci Pollut Res 19:3798–3819
Bressy A, Gromaire M-C, Lorgeoux C, Saad M, Leroy F, Chebbo G (2011) Towards the determination of an optimal scale for stormwater quality management: Micropollutants in a small residential catchment. Water Res 46(20):6799–6810
Bu Q, Wang B, Huang J, Deng S, Yu G (2013) Pharmaceuticals and personal care products in the aquatic environment in China: A review. J Hazard Mat 262:189–211
Cailleaud K, Forget-Leray J, Souissi S, Lardy S, Augagneur S, Budzinski H (2007) Seasonal variation of hydrophobic organic contaminant concentrations in the water-column of the Seine Estuary and their transfer to a planktonic species Eurytemora affinis (Calanoïd, copepod). Part 2: Alkylphenol-polyethoxylates. Chemosphere 70:281–287
Camacho-Muñoz D, Martín J, Santos JL, Aparicio I, Alonso E (2010) Occurrence, temporal evolution and risk assessment of pharmaceutically active compounds in Doñana Park (Spain. J Hazard Mater 183:602–608
Camacho-Muñoz D, Martín J, Santos JL, Aparicio I, Alonso E (2014) Occurrence of surfactants in wastewater: Hourly and seasonal variations in urban and industrial wastewaters from Seville (Southern Spain. Sci Total Environ 468-469:977–984
Campo J, Pérez F, Masiá A, Picó Y, Farré M, Barceló D (2015) Perfluoroalkyl substance contamination of the Llobregat River ecosystem (Mediterraneas area, NE Spain. Sci Total Environ 503:48–57
Castiglioni S, Valsecchi S, Polesello S, Rusconi M, Melis M, Palmiotto M, Manenti A, Davoli E, Zuccato E (2015) Sources and fate of perfluorinated compounds in the aqueous environment and in drinking water of a highly urbanized and industrialized area in Italy. J Hazard Mat 282:51–60
Corada-Fernández C, Jiménez-Martínez J, Candela L, González-Mazo E, Lara-Martín PA (2015) Occurrence and spatial distribution of emerging contaminants in the unsaturated zone. Case study: Guadalete River basin (Cadiz, Spain. Chemosphere 119:S131–S137
Corada-Fernández C, Lara-Martín PA, Candela L, González-Mazo E (2011) Tracking sewage derived contamination in riverine settings by analysis of synthetic surfactants. J Environ Monit 13:2010–2017
da Silva BF, Jelic A, López-Serna R, Mozeto AA, Petrovic M, Barceló D (2011) Occurrence and distribution of pharmaceuticals in surface water, suspended solids and sediments of the Ebro river basin, Spain. Chemosphere 85:1331–1339
Domínguez-Morueco N, González-Alonso S, Valcárcel Y (2014) Phthalate occurrence in rivers and tap water. Sci Total Environ 500–501:139–146
EC (2001) European commission 2001. Decision No 2455/2001/EC of the European Parliament and of the Council of 20 November 2001 establishing the list of priority substances in the field of water policy and amending Directive 2000/60/EC (Text with EEA relevance)
EC (2003) Technical Guidance Document on Risk Assessment in support of Commission Directive 93/67/EEC on Risk Assessment for new noti- fied substances, Commission Regulation (EC) No 1488/94 on Risk Assessment for existing substances, and Directive 98/8/EC of the European Parliament and of the Council concerning the placing of biocidal products on the market. Part II: Environmental Risk Assessment). Office for Official Publications of the European Communities, Luxembourg
EC (2013) Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy. Off. J. Eur. Union L, 226
EC (2015) Commission Implementing Decision (EU) 2015/495 of 20 March 2015 Establishing a Watch List of Substances for Union-wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council
EMEA (2006) Guideline on the environmental risk assessment of medicinal products for human use The European Agency for the Evaluation of Medicinal Products: Committee for Medical Products for Human Use; EMEA/CHMP/SWP/4447/00
Ginebreda A, Muñoz I, López de Alda M, Brix R, López-Doval J, Barceló D (2010) Environmental risk assessment of pharmaceuticals in rivers: Relationships between hazard indexes and aquatic macroinvertebrate diversity indexes in the Llobregat River (NE Spain. Environ Int 36:153–162
Gorga M, Insa S, Petrovic M, Barceló D (2015) Occurrence and spatial distribution of EDCs and related compounds in waters and sediments of Iberian rivers. Sci Total Environ 503–504:69–86
Gros M, Petrovic M, Ginebreda A, Barceló D (2010) Removal of pharmaceuticals during wastewater treatment and environmental risk assessment using hazard indexes. Environ Int 36:15–26
Grujić S, Vasiljević T, Laušević M (2009) Determination of multiple pharmaceutical classes in surface and ground waters by liquid chromatography–ion trap–tandem mass spectrometry. J Chromatogr A 1216:4989–5000
Guerra P, Kim M, Teslic S, Alaee M, Smyth SA (2015) Bisphenol-A removal in various wastewater treatment processes: Operational conditions, mass balance, and optimization. J Environ Manage 152:192–200
Haman C, Dauchy X, Rosin C, Munoz J-F (2015) Occurrence, fate and behavior of parabens in aquatic environments: A review. Water Res 68:1–11
Hernando MD, Mezcua M, Fernández-Alba AR, Barceló D (2006) Environmental risk assessment of pharmaceutical residues in wastewater effluents, surface waters and sediments. Talanta 69:334–342
Huang YQ, Wong CKC, Zheng JS, Bouwman H, Barra R, Wahlström B, Neretin L, Wong MH (2012) Bisphenol A (BPA) in China: a review of sources, environmental levels, and potential human health impacts. Environ Int 42:91–99
Jonkers N, Kohler H-P-E, Dammshäuser A, Giger W (2009) Mass flows of endocrine disruptors in the Glatt River during varying weather conditions. Environ Pollut 157:714–723
Jonkers N, Sousa A, Galante-Oliveira S, Barroso CM, Kohler H-PE, Giger W (2010) Occurrence and sources of selected phenolic endocrine disruptors in Ria de Aveiro, Portugal. Environ Sci Pollut Res 17:834–843
Jurado A, Vàzquez-Suñé E, Carrera J, López de Alda M, Pujades E, Barceló D (2012) Emerging organic contaminants in groundwater in Spain: a review of sources, recent occurrence and fate in a European context. Sci Total Environ 440:82–94
Kuzmanović M, Ginebreda A, Petrović M, Barceló D (2015) Risk assessment based prioritization of 200 organic micropollutants in 4 Iberian rivers. Sci Total Environ 503–504:289–299
Luo Y, Guo W, Ngo HH, Nghiem LD, Hai FI, Zhang J, Liang S, Wang XC (2014) A review on the occurrence of micropollutants in the aquatic environment and their fate and removal during wastewater treatment. Sci Total Environ 473-474:619-641
Martín J, Camacho-Muñoz D, Santos JL, Aparicio I, Alonso E (2011) Monitoring of pharmaceutically active compounds on the Guadalquivir River basin (Spain): occurrence and risk assessment. J Environ Monit 13:2042–2049
Martín J, Camacho-Muñoz D, Santos JL, Aparicio I, Alonso E (2014) Determination of emerging and priority industrial pollutants in surface water and wastewater by liquid chromatography-negative electrospray ionization tandem mass spectrometry Euroanalysis XVII. Anal Bioanal Chem 406:3709–3716
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 75(13):3019–3030
Meffe R, Bustamante I (2014) Emerging organic contaminants in surface water and groundwater: A first overview of the situation in Italy. Sci Total Environ 481:280–295
Mendoza A, Aceña J, Pérez S, López de Alda M, Barceló D, Gil A, Valcárcel Y (2015) Pharmaceuticals and iodinated contrast media in a hospital wastewater: A case study to analyse their presence and characterise their environmental risk and hazard. Environ Res 140:225–241
Moreno-González R, Rodríguez-Mozaz S, Gros M, Pérez-Cánovas E, Barceló D, León VM (2014) Input of pharmaceuticals through coastal surface watercourses into a Mediterranean lagoon (Mar Menor, SE Spain): sources and seasonal variations. Sci Total Environ 490:59–72
Oliveira T, Santacroce G, Coleates R, Hale S, Zevin P, Belasco B (2011) Concentrations of polychlorinated biphenyls in water from US Lake Ontario tributaries between 2004 and 2008. Chemosphere 82:1314–1320
Paíga P, Santos LHMLM, Ramos S, Jorge S, Gabriel Silva J, Delerue-Matos C (2016) Presence of pharmaceuticals in the Lis river (Portugal): Sources, fate and seasonal variation. Sci Total Environ 573:164–177
Papageorgiou M, Kosma C, Lambropoulou D (2016) Seasonal occurrence, removal, mass loading and environmental risk assessment of 55 pharmaceuticals and personal care products in a municipal wastewater treatment plant in Central Greece. Sci Total Environ 543:547–569
Peng X, Yu Y, Tang C, Tan J, Huang Q, Wang Z (2008) Occurrence of steroid estrogens, endocrine-disrupting phenols, and acid pharmaceutical residues in urban riverine water of the Pearl River Delta, South China. Sci Total Environ 397:158–166
Pitarch E, Cervera MI, Portolés T, Ibáñez M, Barreda M, Renau-Pruñonosa A, Morella I, López F, Albarrán F, Hernández F (2016) Monitoring of organic micro-pollutants in surface and groundwater in the surrounding of a solid-waste treatment plant of Castellón, Spain. Sci Total Environ 548-549:211–220
Quednow K, Püttmann W (2008) Endocrine disruptors in freshwater streams of Hesse. Germany: changes in concentration levels in the time span from 2003 to 2005. Environ Pollut 152:476–483
Renz L, Volz C, Michanowicsz D, Ferrar K, Christian C, Lenzner D, El-Hefnawy T (2013) A study of parabens and bisphenol A in surface water and fish brain tissue from the Greater Pittsburgh Area. Ecotoxicology 22:632–641
Robert PH, Thomas KV (2006) The occurrence of selected pharmaceuticals in wastewater effluent and surface waters of the lower Tyne catchment. Sci Total Environ 356(1–3):143–153
Santos JL, Aparicio I, Callejón M, Alonso E (2009) Occurrence of pharmaceutically active compounds during 1-year period in wastewaters from four wastewater treatment plants in Seville (Spain. J Hazard Mat 164:1509–1516
Santos HMLML, Paíga P, Araújo AN, Pena A, Delerue-Matos C, Montenegro BSMMC (2013) Development of a simple analytical method for the simultaneous determination of paracetamol, paracetamol-glucuronide and p-aminophenol in river water. J Chromatogr B 930:75–81
Staniszewska M, Koniecko I, Falkowska L, Burska D, Kiełczewska J (2016) The relationship between the black carbon and bisphenol A in sea and river sediments (Southern Baltic. J Environ Sci 41:24–32
Tauxe-Wuersch A, De Alencastro LF, Grandjean D, Tarradellas J (2005) Occurrence of several acidic drugs in sewage treatment plants in Switzerland and risk assessment. Water Res 39:1761–1772
Terasaki M, Kamata R, Shiraishi F, Makino M (2009) Evaluation of estrogenic activity of parabens and their chlorinated derivatives by using the yeast two-hybrid assay and the enzyme-linked immunosorbent assay. Environ Toxicol Chem 28:204–208
Tijani JO, Fatoba OO, Babajide OO, Petrik LF (2016) Pharmaceuticals, endocrine disruptors, personal care products, nanomaterials and perfluorinated pollutants: a review. Environ Chem Lett 14:27–49
Tran BC, Teil MJ, Blanchard M, Alliot F, Chevreuil M (2015) BPA and phthalate fate in a sewage network and an elementary river of France. Influence of hydroclimatic conditions. Chemosphere 119:43–51
Vazquez-Roig P, Andreu V, Blasco C, Picó Y (2012) Risk assessment on the presence of pharmaceuticals in sediments, soils and waters of the Pego–Oliva Marshlands (Valencia, eastern Spain. Sci Total Environ 440:24–32
Vieno NM, Tuhkanen T, Kronberg L (2006) Analysis of neutral and basic pharmaceuticals in sewage treatment plants and in recipient rivers using solid phase extraction and liquid chromatography–tandem mass spectrometry detection. J Chromatogr A 1134:101–111
Vitaly M, Ensabella F, Stella D, Guidotti M (2004) Nonylphenols in freshwaters of the hydrologic system of an Italian district: association with human activities and evaluation of human exposure. Chemosphere 57:1637–1647
Vorkamp K, Bossi R, Bester K, Bollmann UE, Boutrup S (2014) New priority substances of the European Water Framework Directive: Biocides, pesticides and brominated flame retardants in the aquatic environment of Denmark. Sci Total Environ 470–471:459–468
Wang L, Ying G-G, Zhao J-L, Yang X-B, Chen F, Tao R, Liu S, Zhou L-J (2010) Occurrence and risk assessment of acidic pharmaceuticals in the Yellow River, Hai Riverand Liao River of north China. Sci Total Environ 408:3139–3147
Yamamoto H, Tamura I, Hirata Y, Kato J, Kagota K, Katsuki S, Yamamoto A, Kagami Y, Tatarazako N (2011) Aquatic toxicity and ecological risk assessment of seven parabens: Individual and additive approach. Sci Total Environ 410-411:102–111
Yang Y, Fu J, Peng H, Hou L, Liu M, Zhou JL (2011) Occurrence and phase distribution of selected pharmaceuticals in the Yangtze Estuary and its coastal zone. J Hazard Mat 190:588–596
Zareitalabad P, Siemens J, Hamer M, Amelung W (2013) Perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in surface waters, sediments, soils and wastewater - A review on concentrations and distribution coefficients. Chemosphere 91(6):725–732
Zhou LJ, Ying GG, Zhao JL, Yang JF, Wang L, Yang B, Liu S (2011) Trends in the occurrence of human and veterinary antibiotics in the sediments of the Yellow River, Hai River and Liao River in northern China. Environ Pollut 159:1877–1885
Zolfaghari M, Drogui P, Seyhi B, Brar SK, Buelna G, Dub R (2014) Occurrence, fate and effects of Di (2-ethylhexyl) phthalate in wastewater treatment plants: A review. Environ Pollut 194:281–293
Zúñiga-Benítez H, Peñuela GA (2016) Degradation of ethylparaben under simulated sunlight using photo-Fenton. Water Sci Technol 73:818–826
Acknowledgements
This work has been financed by Ministerio de Economía y Competitividad (project CGL2013-44402-R) Spain.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Electronic supplementary material
ESM 1
(DOCX 6858 kb)
Rights and permissions
About this article

Cite this article
Garrido, E., Camacho-Muñoz, D., Martín, J. et al. Monitoring of emerging pollutants in Guadiamar River basin (South of Spain): analytical method, spatial distribution and environmental risk assessment. Environ Sci Pollut Res 23, 25127–25144 (2016). https://doi.org/10.1007/s11356-016-7759-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-7759-x