
Abstract
Textile manufacturing is the one responsible for water bodies’ contamination through the discharge of colored wastes. This work presents the study of reactive yellow HF (RYHF) dye degradation under two different electrochemical advanced oxidation processes (EAOP), namely anodic oxidation (AO) and electro-Fenton (EF)/boron-doped diamond (BDD) process. For the AO, 100 and 300 mg/L solutions using Pt and BDD as anodes in a 100 mL stirred tank cell were used, with a supporting electrolyte of 0.05 mol/L of Na2SO4 at pH 3 under 30 and 50 mA/cm2 current density. The EF/BDD process was carried out in a flow reactor at 4 and 7 L/min to degrade 100, 200, and 300 mg/L RYHF solutions under 50 and 80 mA/cm2. UV-Vis determinations were used for decolorization evaluation, while high-performance liquid chromatography (HPLC) method provided information on dye degradation rate.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dye is a colored organic structure that intensely absorbs light in the visible region and attaches strongly to fibers through a chemical/physical relationship between the dye and the fiber (Iqbal 2008). Dye compounds are composed of two main components, namely the chromophores that are used to produce color and the auxochromes that give the dye the chemical attraction to the fibers (Gupta et al. 2004). Dyes, which are mainly used to color products of industries such as textile, cosmetic, paper, or leather, are discharged as waste into water effluents, bringing potential hazards to human beings and environment, in general, since some of them are toxic or carcinogenic or even may cause skin and eye irritations. In trying to solve this health/environmental problem, different chemical and physical processes are currently available. Nevertheless, most conventional methods (adsorption on activated carbon, ultrafiltration, and reverse osmosis) are non-destructive, merely transferring pollutants from one phase to another (Gupta et al. 2004; Foo and Hameed 2010; Buscio et al. 2015; Han et al. 2016). Biodegradation has also been tested (Movafeghi et al. 2016), but this method is slow and with low dye degradation activity for most of the strains.
On the other hand, various authors have reported effective treatments for different dyes by means of advanced oxidation processes (AOPs) (Robinson et al. 2001; Brillas and Martínez-Huitle 2015; Sirés et al. 2014). The common characteristic of AOPs is the in situ production of several reactive oxygen species (ROS), mainly the free hydroxyl radical (·OH) with the high standard reduction potential (E° = 2.8 V/SHE), which can oxidize the most hazardous organics to mineralization to CO2, inorganic ions, and water. Recently, electrochemical methods have attracted significant attention for treating recalcitrant toxic wastes containing different dyes (Peralta-Hernández et al. 2009), which are suitable for giving their environmental compatibility, versatility, simplicity, and flexible automation.
The anodic oxidation performance strongly depends on the anode material. Various anode materials have been tested for the degradation of organic compounds; however, the total mineralization with optimal faradic efficiency was obtained by using high oxygen overpotential surfaces, such as SnO2 (Comninellis and Pulgarin 1993; Zanta et al. 2003), PbO2 (Belhadj-Tahar and Savall 1998; Cossu et al. 1998), and especially boron-doped diamond (BDD) (Montilla et al. 2002; Canizares et al. 2004; Boye et al. 2002; Cruz-González et al. 2012). The anodic oxidation with BDD anodes is considered among the most efficient processes. The generation of the physisorbed oxidant BDD(·OH) in the BDD anode is written as follows:
Another widely used electrochemical AOP (EAOP) is the electro-Fenton process (EFP) (Moreira et al. 2013; Lahkimi et al. 2007; Garcia-Segura et al. 2011). It consists of a continuous cathodic production of H2O2 via O2 reduction (reaction (2)) with air injection into the treated solution or directly on a carbonaceous cathode. Small amounts of iron Fe2+ are added to the medium to react with H2O2, generating Fe3+ and ·OH in the bulk from Fenton’s reaction (3), with an optimum pH of about 2.8–3.0. This reaction is catalytic since it can be propagated from Fe2+ regeneration by cathodic Fe3+ reduction (Panizza and Cerisola 2009).
Carbon nanotubes (Khataee et al. 2013), graphite (Rivera et al. 2011), activated carbon fiber (Wang et al. 2008), carbon sponge (Daneshvar et al. 2008), graphite felt (Panizza and Oturan 2011), carbon felt (El-Ghenymy et al. 2014; Sopaj et al. 2015), and carbon-polytetrafluoroethylene (PTFE) O2 or air diffusion (Flox et al. 2007; Florenza et al. 2014) electrodes have been used for the efficient H2O2 production from reaction (2). More recently, there has been an increased interest in the BDD material (Cruz-González et al. 2010; Cruz-González et al. 2012; Uranga-Flores et al. 2015), giving the H2O2 production when used in Fenton-type processes:
In addition to the above discussion, it is important to state that the electrochemical advanced oxidation processes are also strongly influenced by the origin/type of organic compounds to be treated, as it was demonstrated recently by Garcia-Segura et al., where they studied the degradation of two different azo dyes by means of electro-Fenton processes.
The present study was stimulated by the authoritative paper research on decontamination of reactive yellow 160 azo dye by electrochemical methods published by Bedolla-Guzman et al. 2016; in this paper, the authors performed a comparative study of anodic oxidation with electrogenerated H2O2 (AO-H2O2), electro-Fenton (EF), and UVA photoelectro-Fenton (PEF) to a degraded constant azo dye concentration of 0.167 mmol dm−3 using 100 cm3 of a solution.
Now, in the current paper, we present the results from decolorization and degradation of the reactive yellow HF (RYHF) azo dye solutions when applying anodic oxidation and electro-Fenton processes. Comparative trials were made by providing a constant current density with a power supply. The effect of the current applied and flow rate on the degradation was examined to clarify the role of generated ·OH. The dye decolorization was followed by UV-Vis determinations. Reductions of azo dye concentrations were quantified by HPLC chromatography.
In the light of this proposal and as natural extension to improve our early results, we get into the following questions: Could it be possible to succeed in the RYHF azo dye degradation using Pt or BDD anode? Which could be the effect from the flow rate?
Experimental
Chemicals
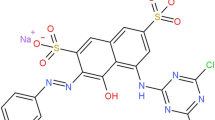
Commercial RYHF (66 % content, the other components were inorganic salts for stabilization) was supplied by PCL S.A de C.V (Mexico) (C25H22ClN9Na2O12S3; Table 1). The solutions were prepared with ultrapure water from a Millipore Milli-Q system (resistivity >18 MΩ cm, 25 °C). The catalyst and background electrolyte were introduced as FeSO4·7H2O and anhydrous Na2SO4, both of the analytical grades were purchased from Karal and JT. Baker, respectively. The solution’s pH was adjusted to 2.8–3.0 with analytical grade H2SO4 supplied by Merck. Solvents and other chemicals were either of analytical or HPLC grade purchased from Panreac and Sigma-Aldrich.
Electrochemical system setup
The first part of the experiments was carried out in a stirred tank cell at laboratory scale. In all assays, the solution was vigorously stirred with a magnetic bar at 500 rpm in order to mix the organics and transport their mass toward/from the electrodes, see Fig. 1. The electrodes were a 5-cm2 BDD thin film purchased from Metakem™ Germany and 3 cm2 Pt purchased from Electrochem™. The inter-electrode gap was of about 1 cm. All the trials were made at a constant current provided by a BK precision 1627 A power supply. Solutions with 100 and 300 mg/L of RYHF in 0.05 mM Na2SO4 were degraded at pH 2.8–3.0 by AO/Pt and AO/BDD. The influence of the current applied between 30 and 50 mA/cm2 on the performance of each EAOP was evaluated.

Scheme of the experimental setup used for the anodic oxidation treatments. Stirred electrolytic cell containing the treated RYHF solution, magnetic stirred bar, stainless steel cathode, platinum or BDD anode, and power supply
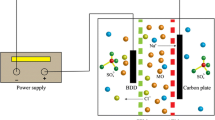
The second set of electrochemical experiments was implemented at room temperature in a 3-L undivided filter flow press reactor prepared with two electrodes, see Fig. 2. The anode and cathode electrodes had a geometrical area of 64 cm2 of niobium plate with a 2–7-μm-thick diamond-coated film. BDD electrodes were provided by Metakem GmbH™, Germany. The inter-electrode gap was of 5 mm. The synthetic solutions were placed in the tank and recirculated using a diaphragm pump (Ramírez et al. 2013). The flow rate was kept at 4 and 7 L/min. The degradation of 100, 200, and 300 mg/L of aqueous synthetic dye RYHF solutions was carried out under current controlled electrolysis conditions at 50 and 30 mA/cm2 using a BK Precision 1688B power supply.

Experimental laboratory pilot plan used for RYHF electro-Fenton process. The electrochemical reactor was a one-compartment BDD/BDD filter press cell
Procedures
The decolorization of RYHF solutions was monitored from their absorbance (A) removal at the maximum wavelength of 450 nm in the visible region, determined on a Cintra 1010 UV-Vis spectrophotometer. The decolorization rate or percentage of color removal for each sample was calculated from Eq. (5) (Ruiz et al. 2011; Brillas and Martínez-Huitle, 2015; Bedolla et al. 2016):
where A 0 and A t are the initial and final adsorbance times, respectively, at λmax = 450 nm.
For the optimized EAOP assays, the RYHF concentration removal was monitored by reversed-phase high-performance liquid chromatography (HPLC) with an Agilent 1260 infinity, fitted with an Agilent Eclipse C18 PAH reverse phase (250 mm × 4.6 mm × 3 mm particle size) and coupled with UV detector. Aliquots of 20 μL were injected and a 30:30:40 (v/v) CH3OH/H2O/CH3CN mixture was eluted at 0.8 mL/min as mobile phase. The chromatograms displayed a peak for RYHF anion with a retention time (tr) of 3.9 min.
Results and discussion
AO treatment of RYHF solutions
The first decolorization of RYHF solutions was carried out by anodic oxidation using a stirred tank cell. This electrochemical technique destroys pollutants mainly by the action of the ·OH formed as intermediaries on a high O2-overvoltage anode surface from water oxidation, using Pt and BDD as anode materials. A solution of 100 and 300 mg/L of RYHF at pH 3.0 was electrolyzed to test its comparative degradation through the anodic oxidation of the Pt and BDD anodes.
In both cases, the initial yellow solution turned brown at 20 min of treatment due to the formation of some soluble aromatic products. After 120 min of electrolysis using the Pt or BDD, color intensity gradually decreased to obtain a less colored solutions anode. Figure 3a shows the change in the color removal (%) with 30 and 50 mA/cm2 of current intensity and 100 and 300 mg/L of dye concentration for Pt anode. It is possible to observe that an increase in the current density improves the decolorization rate. With a 30 mA/cm2 current and 100 mg/L RYHF (•), low decolorization is observed, with only 30 %. When the RYHF concentration is increased to 300 mg/L (▼), there is decolorization of almost 40 %. Increasing the current to 50 mA/cm2, the decolorization is of 50 % and with 300 mg/L of dye (♦), while the highest dye concentration, a 62 % of decolorization is reached (■). However, this result demonstrated the low ability of the Pt anode to decolorize the azo dye compound, given that the ·OH is formed on the Pt surface and adsorbed on the metal, which considerably decreases the degradation capacity (Flox et al. 2005).

Variation of percentage of color removal a Pt anode and b BDD anode, using a laboratory stirred tank cell for the degradation of 100 mL of solutions at pH 3.0 containing 100 mg/L of RYHF at current intensity of 30 mA/cm2 (filled circle) and 50 mA/cm2 (inverted filled triangle); 300 mg/L RYHF at current intensity of 30 mA/cm2 (filled diamond) and 50 mA/cm2 (filled square)
Better color abatement can be observed for BDD anode (Fig. 3b). With 30 mA/cm2 current intensity and 100 mg/L of dye, 37 % was reached (•). When current raised to 50 mA/cm2 (▼), 57 % was obtained. Sixty percent (♦) was reached with 300 mg/L and the current density of 30 mA/cm2. The best result was obtained with 50 mA/cm2 and 300 mg/L, where 70 % of the decolorization was achieved (■). The higher decolorization rate with increasing current and dye concentration can be simply associated with the concomitant acceleration of reaction (1), thereby generating larger quantities of BDD(·OH) that destroy more rapidly the conjugated chromophore system of RYHF, when such aromatic species were completely destroyed by the ·OH produced on the surface of each anode. The above demonstrates the existence of a complex decolorization behavior of RYHF for both electrodes. Given that current density and the dye concentration play a crucial role, a chromatographic study was carried out to learn more about decreased concentrations on both electrodes. Figure 4 illustrates the evolution of RYHF degradation by anodic oxidation with Pt (•) and BDD (♦) anodes, respectively, under HPLC analysis. This study was carried out with a 50 mA/cm2 current and 300 mg/L of dye concentration for both anodes during 120 min, obtaining 36 % of degradation with the Pt and 70 % with the BDD.

Evolution of degradation percentage rate for 300 mg/L of RYHF at 50 mA/cm2 current intensity in a 100 mL solution using a laboratory stirred tank cell under HPLC analysis; Pt anode (filled circle), BDD anode (filled diamond)
The above results are indicative of a fast degradation of RYHF and its products by anodic oxidation with BDD, which are feasible by the efficient generation of ·OH on the anode surface from reaction (1). However, the use of a Pt anode under comparable conditions leads to a poor depollution because it produces smaller amounts of highly reactive ·OH on its surface to destroy organics.
EF/BDD treatment of RYHF solutions
Several assays for the decolorization of the solutions containing RYHF by the EF/BDD process were performed at the 3-L undivided filter flow press reactor. Figure 5 illustrates the decolorization rate for the electrolysis with 50 and 80 mA/cm2 current intensity to treat 100, 200, and 300 mg/L of RYHF solution with pH 3 at a flow rate of 4 L/min and adding 0.5 mM of Fe2+ during 180 min at room temperature (25 °C). The results (Fig. 5a) also show that RYHF is quickly decolorized during the first 100 min under 50 mA/cm2 current intensity, yielding 83 % of decolorization after 180 min of the treatment when using high concentration of 300 mg/L (▼), whereas at the same time, the use of 200 and 100 mg/L lead to 72 % (♦) and 63 % (•) decolorization, respectively. The use of the EF/BDD filter flow press reactor (by oxygen reduction at BDD cathode surface (reaction 4)) could generate both H2O2 continually and also ·OH in the bulk solution from Fenton’s type reaction (3) by EF processes (Cruz-González et al. 2010). Figure 5b shows that for the higher current intensity, there was a larger loss of color until 180 min of electrolysis, whereupon it was decelerated up to attain decolorization efficiencies of 68 % (•), 81 % (♦), and 90 % (▼) for 80 mA/cm2, respectively. This is indicative of the generation of very recalcitrant colored products that need longer time to be removed by EF/BDD. The production of great amounts of ·OH, with much higher oxidation ability to remove the azo dye and its colored products in EF, gave rise to total decolorization in about 180 min for all current intensity values.

Percentage of color removal for EF/BDD process carry out in a flow reactor at 4 L/min flow rate and 3 L solution a using 50 mA/cm2 current intensity for 100 mg/L (filled circle), 200 mg/L (filled diamond), and 300 mg/L (inverted filled triangle) RYHF solutions; and b using 80 mA/cm2 current intensity for 100 mg/L (filled circle), 200 mg/L (filled diamond), and 300 mg/L (inverted filled triangle) RYHF solutions
Figure 6a illustrates the decolorization rates for the electrolysis of a 100 (•), 200 (♦), and 300 mg/L (▼) RYHF solution at 7 L/min, 50 mA/cm2 current intensity, and adding 0.05 mM Fe2+. The results showed a similar behavior in the case of the 4 L/min flow rates tested (Fig. 5), that is to say, removal rates close to 85 % for all three concentrations tested. A slight improvement is obtained when the current intensity is increased to 80 mA/cm2 (Fig. 6b) with concentrations of 100 and 200 mg/L, and after 100 min, 73 % decolorization is achieved (•, ♦), and with 300 mg/L 81 %, and at the end of the process, 91 % decolorization is obtained (▼), which represents the best result obtained in this set of studies. When EF/BDD was applied, the strong yellow color lost intensity turning into dark brown that slowly became colorless. This suggests that the gradual production of colored conjugated products was slowly destroyed mainly by ·OH formed in the bulk from Fenton’s reaction (3).

Percentage of color removal for EF/BDD process carry out in a flow reactor at 7 L/min flow rate and 3 L solution a using 50 mA/cm2 current intensity for 100 mg/L (filled circle), 200 mg/L (filled diamond), and 300 mg/L (inverted filled triangle) RYHF solutions; and b using 80 mA/cm2 current intensity for 100 mg/L (filled circle), 200 mg/L (filled diamond), and 300 mg/L (inverted filled triangle) RYHF solutions
These results show that an increase in the flow rate virtually does not improve decolorization as indicated by the curves in Figs. 5 and 6. Additionally, in order to evaluate the effects of the flow rate in the abatement of the azo RYHF concentration, reversed phase liquid chromatography tests were performed. These chromatograms exhibited a well-defined peak for this compound at tr = 3.9 min. As shown in Fig. 7a, when the 4 L/min flow rate is applied, under 50 mA/cm2 current intensity (•) to degrade 300 mg/L of RYHF, it is possible to observe an important increase in the degradation rate, where in the first 100 min, the process achieved 81 % of degradation and 90 % at the end of the treatment. For the case with 80 mA current intensity (♦), the higher degradation is obtained; only in the first 100 min the dye degraded to 92 %. A gradual increase in RYHF removal is caused by the rising current from 50 to 80 mA/cm2 in both cases. This enhancement of the degradation can be associated with a faster destruction of pollutants by the increase in rate of reaction (3) to produce more quantity of ·OH, as well as the accumulation of more H2O2 from reaction (4), where the effect of the current intensity is evident.

Evolution of degradation percentage rate for 300 mg/L of RYHF in a 3 L solution using a flow reactor under HPLC analysis. a 4 L/min at 50 mA/cm2 current intensity (filled circle) and 80 mA/cm2 current intensity (filled diamond); b 7 L/min at 50 mA/cm2 current intensity (filled circle) and 80 mA/cm2 current intensity (filled diamond)
An interesting result is shown in Fig. 7b, corresponding to the RYHF degradation at 7 L/min flow rate. It can be seen that for both current intensities 50 and 80 mA/cm2 (•, ♦), there is no obvious improvement in the dye degradation since in the first few minutes, the degradation evolves similar values but toward the end of the treatment, only 98 % degradation is obtained. This result is an important evidence that some increment in the flow rate is a positive influence on the dye degradation rate; a possible explanation is that the rate of mass transfer is favored at high flow regimes; however, more studies are still needed to better understand the phenomenon.
In light of these results, it is important to note that authors such as Garcia-Segura and Brillas (2014) and Garcia-Segura and Brillas (2016) have already shown that the use of reactors at the pilot level to degrade various organic compounds via the electrochemical advanced oxidation processes are viable instruments that allow studies greater volumes of solution, as well as to know the influence of the most important variables such as flow rate, current density, concentration of the compound, and to demonstrate the effectiveness of this type of technologies with a view of their application in real processes.
Conclusions
The obtained results demonstrate that anodic oxidation and electro-Fenton with a BDD anode are viable methods to treat acidic wastewaters containing RYHF. The ·OH formed from Fenton’s reaction (3) removes more rapidly aromatic products, thus, making the electro-Fenton process more efficient than anodic oxidation. The alternative use of a Pt anode in the same methods under comparable conditions leads to slower decolorization given the lower oxidizing power of Pt(·OH). On the other hand, it is important to highlight that the increase in current intensity accelerates the decolorization of all pollutants, but with the consumption of more specific charge. The flow rate has little influence on the decolorization rate for the anodic oxidation; however, in the case of EF/BDD process, a considerable improvement can be observed, achieving almost 100 % of degradation.
References
Bedolla-Guzman A, Sirés I, Thiam A, Peralta-Hernández JM, Gutiérrez-Granados S, Brillas E (2016) Application of anodic oxidation, electro-Fenton and UVA photoelectro-Fenton to decolorize and mineralize acidic solutions of Reactive Yellow 160 azo dye. Electrochim Acta 206:307–316
Belhadj-Tahar N, Savall A (1998) Mechanistic aspects of phenol electrochemical degradation by oxidation on a Ta/PbO2 anode. J Electrochem Soc 145:3427–3434
Boye B, Michaud PA, Marselli B, Dieng MM, Brillas E, Comninellis C (2002) Anodic oxidation of 4-chlorophenoxyacetic acid on synthetic boron-doped diamond electrodes. New Diamond Front Carbon Technol 12:63–72
Brillas E, Martínez-Huitle CA (2015) Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods. An updated review. Appl Catal B Environ 166-167:603–643
Buscio V, Marín MJ, Crespi M, Gutiérrez-Bouzán C (2015) Reuse of textile wastewater after homogenization-decantation treatment coupled to PVDF ultrafiltration membranes. Chem Eng J 265:122–128
Canizares P, Garcia-Gomez J, Saez C, Rodrigo MA (2004) Electrochemical oxidation of several chlorophenols on diamond electrodes: part II. Influence of waste characteristics and operating conditions. J Appl Electrochem 34:87–94
Comninellis C, Pulgarin C (1993) Electrochemical oxidation of phenol for wastewater treatment using SnO2, anodes. J Appl Electrochem 23:108–112
Cossu R, Polcaro AM, Lavagnolo MC, Mascia M, Palmas S, Renoldi F (1998) Electrochemical treatment of landfill leachate: oxidation at Ti/PbO2 and Ti/SnO2 anodes. Environ Sci Technol 32:3570–3573
Cruz-González K, Torres-López O, García-León A, Guzmán-Mar JL, Reyes LH, Hernández-Ramírez A, Peralta-Hernández JM (2010) Determination of optimum operating parameters for Acid Yellow 36 decolorization by electro-Fenton process using BDD cathode. Chem Eng J 160:199–206
Cruz-González K, Torres-Lopez O, García-León AM, Brillas E, Hernández-Ramírez A, Peralta-Hernández JM (2012) Optimization of electro-Fenton/BDD process for decolorization of a model azo dye wastewater by means of response surface methodology. Desalination 286:63–68
Daneshvar N, Aber S, Vatanpour V, Rasoulifard MH (2008) Electro-Fenton treatment of dye solution containing Orange II: influence of operational parameters. J Electroanal Chem 615:165–174
El-Ghenymy A, Rodríguez RM, Brillas E, Oturan N, Oturan MA (2014) Electro-Fenton degradation of the antibiotic sulfanilamide with Pt/carbon-felt and BDD/carbon-felt cells. Kinetics, reaction intermediates, and toxicity assessment. Environ Sci Pollut Res 21:8368–8378
Florenza X, Solano AMS, Centellas F, Martínez-Huitle CA, Brillas E, Garcia-Segura S (2014) Degradation of the azo dye Acid Red 1 by anodic oxidation and indirect electrochemical processes based on Fenton’s reaction chemistry. Relationship between decolorization, mineralization and products. Electrochim Acta 142:276–288
Flox C, Garrido JA, Rodríguez RM, Cabot PL, Centellas F, Arias C, Brillas E (2007) Mineralization of herbicide mecoprop by photoelectro-Fenton with UVA and solar light. Catal Today 129:29–36
Flox C, Garrido JA, Rodríguez RM, Centellas F, Cabot PL, Arias C, Brillas E (2005) Degradation of 4,6-dinitro-o-cresol from water by anodic oxidation with a boron-doped diamond electrode. Electrochim Acta 50:3685–3692
Foo KY, Hameed BH (2010) An overview of dye removal via activated carbon adsorption process. Desalen Water Treat 19:255–274
Garcia-Segura S, Brillas E (2014) Advances in solar photoelectro-Fenton: decolorization and mineralization of the Direct Yellow 4 diazo dye using na autonomous solar pre-pilot plant. Electrochim Acta 140:384–395
Garcia-Segura S, Brillas E (2016) Combustion of textile monoazo, diazo and triazo dyes by solar photoelectron-Fenton: decolorization, kinetics and degradation routes. Appl Catal B Environ 181:681–691
Garcia-Segura S, Centellas F, Arias C, Garrido JA, Rodríguez RM, Cabot PL, Brillas E (2011) Comparative decolorization of monoazo, diazo and triazo dyes by electro-Fenton process. Electrochim Acta 58:303–311
Gupta VK, Suhas AI, Saini VK (2004) Removal of rhodamine B fast green and methylene blue from wastewater using red mud an aluminum industry waste. Ind Eng Chem Res 43:1740–1747
Han G, Liang CZ, Chung TS, Weber M, Staudt C, Maletzko C (2016) Combination of forward osmosis (FO) process with coagulation/flocculation (CF) for potential treatment of textile wastewater. Water Res 91:361–370
Iqbal M (2008) Textile dyes. Rehbar, Karachi
Khataee A, Khataee A, Fathinia M, Vahid B, Joo SW (2013) Kinetic modeling of photoassisted-electrochemical process for degradation of an azo dye using boron-doped diamond anode and cathode with carbon nanotubes. J Ind Eng Chem 19:1890–1894
Lahkimi A, Oturan MA, Oturan N, Chaouch M (2007) Removal of textile dyes from water by the electro-Fenton process. Environ Chem Lett 5:35–39
Montilla F, Michaud PA, Morallon E, Vazquez JL, Comninellis C (2002) Electrochemical oxidation of benzoic acid at boron-doped diamond electrodes. Electrochim Acta 47:3509–3513
Moreira FC, Garcia-Segura S, Vilar VJP, Boaventura RAR, Brillas E (2013) Decolorization and mineralization of Sunset Yellow FCF azo dye by anodic oxidation, electro-Fenton, UVA photoelectro-Fenton and solar photoelectro-Fenton processes. Appl Catal B Environ 142-143:877–890
Movafeghi A, Khataee AR, Moradi Z, Vafaei F (2016) Biodegradation of direct blue 129 diazo dye by Spirodela polyrrhiza: an artificial neural networks modeling. Int J Phytoremediation 18:337–347
Panizza M, Cerisola G (2009) Electro-Fenton degradation of synthetic dyes. Water Res 43:339–344
Panizza M, Oturan MA (2011) Degradation of Alizarin Red by electro-Fenton process using a graphite-felt cathode. Electrochim Acta 56:7084–7087
Peralta-Hernández JM, Martínez-Huitle CA, Guzmán-Mar JL, Hernández-Ramírez A (2009) Recent advances in the application of electro-Fenton and photoelectro-Fenton process for removal of synthetic dyes in wastewater treatment. J Environ Eng Manag 19:257–265
Ramírez C, Saldaña A, Hernández B, Acero R, Guerra R, Garcia-Segura S, Brillas E, Peralta-Hernández JM (2013) Electrochemical oxidation of methyl orange azo dye at pilot flow plant using BDD technology. J Ind Eng Chem 19:571–579
Rivera M, Pazos M, Sanromán MA (2011) Development of an electrochemical cell for the removal of Reactive Black 5. Desalination 274:39–43
Robinson T, McMullan G, Marchant R, Nigam P (2001) Remediation of dyes in textile effluent: a critical review on current treatment technologies with a proposed alternative. Bioresour Technol 77:247–255
Ruiz EJ, Hernández-Ramírez A, Peralta-Hernández JM, Arias C, Brillas E (2011) Application of solar photoelectro-Fenton technology to azo dyes mineralization: effect of current density, Fe2+ and dye concentration. Chem Eng J 171:385–392
Sirés I, Brillas E, Oturan MA, Rodrigo MA, Panizza M (2014) Electrochemical advanced oxidation processes: today and tomorrow. A review. Environ Sci Pollut Res 21:8336–8367
Sopaj F, Rodrigo MA, Oturan N, Podvorica FI, Pinson J, Oturan MA (2015) Influence of the anode materials on the electrochemical oxidation efficiency. Application to oxidative degradation of the pharmaceutical amoxicillin. Chem Eng J 262:286–294
Uranga-Flores A, de la Rosa-Juarez C, Gutierrez-Granados S, Chianca de Moura D, Martinez-Huitle CA, Peralta-Hernandez JM (2015) Electrochemical promotion of strong oxidants to degrade Acid Red 211: effect of supporting electrolytes. J Electroanal Chem 738:84–91
Wang A, Qu J, Liu H, Ru J (2008) Mineralization of an azo dye Acid Red 14 by photoelectro-Fenton process using an activated carbon fiber cathode. Appl Catal B Environ 84:393–399
Zanta CLPS, Michaud PA, Comninellis C, Andrade ARD, Boodts JFC (2003) Electrochemical oxidation of p-chlorophenol on SnO2–Sb2O5 based anodes for wastewater treatment. J Appl Electrochem 33:1211–1215
Acknowledgments
Financial support from PRODEP-UGTO-PTC-472, UGTO under the Project 007/2015 (Convocatoria Institucional para Forltalecer la Exelencia Académica 2015) and Project 778/2016 (Convocatoria Institucional de Apoyo a la Investigación Científica 2016-2017) is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: VÃtor Pais Vilar
Rights and permissions
About this article

Cite this article
Bedolla-Guzman, A., Feria-Reyes, R., Gutierrez-Granados, S. et al. Decolorization and degradation of reactive yellow HF aqueous solutions by electrochemical advanced oxidation processes. Environ Sci Pollut Res 24, 12506–12514 (2017). https://doi.org/10.1007/s11356-016-7286-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-7286-9