
Abstract
Fe2O3 and CeO2 modified activated coke (AC) synthesized by the equivalent-volume impregnation were employed to remove elemental mercury (Hg0) from simulated flue gas at a low temperature. Effects of the mass ratio of Fe2O3 and CeO2, reaction temperature, and individual flue gas components including O2, NO, SO2, and H2O (g) on Hg0 removal efficiency of impregnated AC were investigated. The samples were characterized by Brunauer–Emmett–Teller (BET), X-ray diffraction (XRD), scanning electron microscopy (SEM), and X-ray photoelectron spectroscopy (XPS). Results showed that with optimal mass percentage of 3 % Fe2O3 and 3 % CeO2 on Fe3Ce3/AC, the Hg0 removal efficiency could reach an average of 88.29 % at 110 °C. Besides, it was observed that O2 and NO exhibited a promotional effect on Hg0 removal, H2O (g) exerted a suppressive effect, and SO2 showed an insignificant inhibition without O2 to some extent. The analysis of XPS indicated that the main species of mercury on used Fe3Ce3/AC was HgO, which implied that adsorption and catalytic oxidation were both included in Hg0 removal. Furthermore, the lattice oxygen, chemisorbed oxygen, and/or weakly bonded oxygen species made a contribution to Hg0 oxidation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mercury is a global pollutant because of its toxicity, mobility, and bioaccumulation in the ecosystem and food chain (Pavlish et al. 2003). The emission of mercury from anthropogenic sources is a serious concern that attracts considerable public attention. Coal-fired power plants are considered to be a major source of anthropogenic mercury emissions. By April 2010, more than 20 states in the USA had issued mercury emission regulations to regulate mercury emissions from coal-fired power plants which were more stringent than the Clean Air Mercury Rule (CAMR) (Milford and Pienciak 2009). In order to control mercury emission from coal-fired power plants, the Mercury and Air Toxics Standards (MATS) announced in 2011 by the US Environment Protection Agency (USEPA) emphasized the control of mercury, acid gases, and other toxic pollutants (Zhang et al. 2014). Therefore, it is an extremely urgency to find effective technologies to control mercury emission.
Mercury in coal-fired flue gas is generally presented in three forms, namely elemental mercury (Hg0), oxidized mercury (Hg2+), and particle-bound mercury (Hgp) (Galbreath and Zygarlicke 2000)). Hgp and Hg2+ are relatively easy to be removed by typical air pollution control devices (APCDs). For instance, Hgp can be captured by particulate matter (PM) control devices such as electrostatic precipitators (ESPs) and fabric filters (FFs). Hg2+ is soluble in water, and it can be removed efficiently by wet flue gas desulfurization equipments (WFGDs) (Hsi et al. 2010). However, Hg0 can hardly be removed by currently available APCDs for its high volatility and nearly insolubility in water, which becomes the biggest obstacle for mercury control.
Up to now, sorbents/catalysts like carbon-based sorbents, selective catalytic reduction (SCR) catalysts, and metals or metal oxides (Cao et al. 2007; Presto and Granite 2006) have been used for Hg0 removal. Among them, Hg0 removal with activated carbon has been proved to be an effective technique in commercial application by many researchers (Li et al. 2003). However, the activated carbon owns a distinct disadvantage of high cost. Activated coke (AC) is a kind of porous activated carbon-based sorbent which is not adequately activated. With high mechanical strength, AC could endure attrition and comminution during circulation and handling processes (Jastrząb 2012). AC in granular form is suited to be used in both moving bed and fixed-bed unit. Meanwhile, AC inherits the virtues of activated carbon such as the structure characteristics, adsorption and catalytic properties, physical and chemical stability (Li et al. 2013). In addition, AC can suffice for handling lots of pollutants such as H2S, SO2, NH3, and Hg in exhaust gas under the corresponding temperature (Itaya et al. 2009; Ogriseck and Vanegas 2010; Shawwa et al. 2001).
Due to high volatility and weak bonding onto carbon surface of Hg0, suitable chemical modifications on AC are sought to boost its adsorption and catalytic performance for Hg0 removal. Recently, various transition-metal oxides including Fe2O3, V2O5, CuO, CeO2, MnO2, Mn2O3, Cr2O3, and RuO2 had been widely investigated as potential Hg0 oxidation catalysts, and these metal oxides were valid for oxidizing Hg0 to Hg2+ (Dunham et al. 2003; Fan et al. 2012; Ji et al. 2008; Kamata et al. 2009; Pitoniak et al. 2005; Yan et al. 2011). Thereinto, CeO2 plays an important role in a large number of catalytic reactions, and it has been extensively studied as an oxygen provider by storing or releasing O2 via the unique Ce4+/Ce3+ redox couple (Kaspar et al. 1999). Owing to the redox shift between Ce3+ and Ce4+, labile oxygen vacancies and bulk oxygen species with relatively high mobility can be easily formed (Reddy et al. 2003), which make them active for oxidation process. Thus, CeO2 has been successfully applied to important processes such as catalytic wet oxidation and CO oxidation (Ayastuy et al. 2006; Delgado et al. 2006). Additionally, Kinya et al. reported that the impregnation of Fe on the active carbon greatly enhanced the removal of both COS and H2S at 300–450 °C (Sakanishi et al. 2005). In Fe-Mn mixed oxides, iron cations are replaced by manganese cations and an equivalent number of cation vacancies are incorporated to maintain the spinel structure (Gillot et al. 1997). Yue et al. (Yue and Zhang 2009) reported that incorporation of Fe3+ into fluorite structure of CeO2 induces oxygen-storage systems and can give rise to the increase of oxide ion vacancies which are responsible for their high solid-state ionic conductivity. Consequently, CeO2 coupled with Fe2O3 modified AC may have high removal efficiency for Hg0. To our knowledge, there is little information about Fe2O3 and CeO2 modified AC for Hg0 removal.
In this study, AC is used as a supporter to synthesize Fe2O3-CeO2/AC samples by the equivalent-volume impregnation method. Different operation parameters like reaction temperature and individual flue gas components on Hg0 removal were studied and optimized. Various characterization methods were applied to study the structure and physicochemical characteristic of samples. The ultimate goal of this study is to develop an economical sorbent which is promising for industrial application in Hg0 removal from coal-fired flue gas.
Experimental section
Samples preparation
The AC (columnar granules with a length of 7–9 mm and an average diameter of 5 mm) used in the experiment was obtained from Inner Mongolia Kexing Carbon Industry Limited Liability Company. The samples were prepared by the equivalent-volume impregnation method as follows: At first, AC was washed with deionized water for several times and dried in an electric blast oven at 105 °C for 12 h. After that, different amount of Ce(NO3)3·6H2O and Fe(NO3)3·9H2O were dissolved in deionized water to form mixed solution. Then, a certain amount of AC was impregnated in the solution for 18 h. After impregnation, the samples were dried in an electric blast oven at 105 °C for 12 h and calcinated in an electric tube furnace at 450 °C for 4 h under N2 atmosphere. Finally, the samples were cooled down to room temperature and stored in a desiccator for further use. Fe2O3-CeO2/AC samples were denoted as FexCey/AC, where Fe represents Fe2O3, Ce represents CeO2, x and y represent the mass percentage of Fe2O3 and CeO2 on the total sample mass including AC, Fe2O3, and CeO2, respectively. In the entire experimental procedure, the total mass percentage of Fe2O3 and CeO2 on all samples was 6 %, which was based on our previous work (Xie et al. 2015). Meanwhile, Fe/AC and Ce/AC were prepared with the same method mentioned above.
Samples characterization
Brunauer–Emmett–Teller (BET) was performed by using a TriStarII3020 analyzer (Mcpicromeritics Instrument Corp, USA) to determine the specific surface area and porosity of samples. All of the samples were degassed at 180 °C for 5 h prior to BET measurements.
X-ray diffraction (XRD) measurements were carried out on a Bruker D8-Advance device to examine the crystallinity and dispersivity of iron and cerium species on AC surface using CuKa radiation (λ = 0.1543 nm) in the range of 10–80° (2θ) with a step size of 0.02°.
To further analyze the morphology and surface structure of the samples, scanning electron microscopy (SEM) photographs were obtained by means of a HITACHI S-4800 analyzer.
X-ray photoelectron spectroscopy (XPS) analysis was carried out on a K-Alpha 1063 X-ray photoelectron spectrometer (Thermo Fisher Scientific, USA) with an Al Ka X-ray source at room temperature. The binding energies were calibrated by the C 1 s peak at 284.6 eV.
Experimental setup and procedure
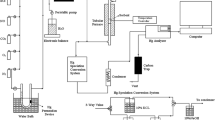
Figure 1 shows the schematic diagram of the experimental system, which was consisted of a simulated flue gas system, Hg0 vapor-generating device, electric tube furnace reactor, and an online mercury analyzer. The simulated flue gas (SFG) contained 70 μg/m3 Hg0, 5 % O2, 8 % H2O (when used), 300 ppm NO, 400 ppm SO2, 12 % CO2, and balanced N2. A flow of high-purity nitrogen (200 ml/min) was used as Hg0-laden gas stream by passing through the Hg0 permeation tube (VICI Metronics). To guarantee a constant permeation concentration, the Hg0 permeation tube was placed in a U-shaped quartz tube, which was immersed in an electric-heated thermostatic water bath. The total flow rate was controlled at 1 L/min in each test using mass flow controllers, corresponding to a space velocity of about 4200 h−1. The electric tube furnace was comprised of a quartz tube (a length of 95 cm and an inner diameter of 52 mm) and matched installations for regulating the reaction temperature. The inlet and outlet Hg0 concentrations were measured by the mercury analyzer (Lumex RA-915 M, Russia). The exhaust gas from the mercury analyzer was introduced into an activated carbon trap before released into the atmosphere.

Schematic diagram of the experimental setup
Five sets of experiments were carried out and the reaction conditions are listed in Table 1. Set 1 experiments were aimed at determining the optimal mass ratio of Fe2O3 and CeO2. The Hg0 removal activity over virgin AC and impregnated AC was evaluated under SFG for 3 h. In Set 2, the optimal sample was tested in SFG at each selected reaction temperature for investigating the optimal reaction temperature. The roles of individual flue gas components on Hg0 removal and the reaction pathways were explored in Set 3 experiments, which were conducted on the optimal sample in the presence of individual flue gas (balanced with N2 or O2 plus N2) at optimal operating temperature. In Set 4 experiments, the optimal sample was tested in SFG for 14 h at optimal operating temperature for studying its durability. Set 5 experiments investigated the breakthrough curves of Hg0 captured by the optimal sample under N2 and N2 plus O2.
During each experiment, the flue gas firstly bypassed the fix-bed reactor until a stable inlet concentration of Hg0 was obtained. When the mercury analyzer had been stable for more than 30 min, the gas flow was switched to pass through the sample and Hg0 concentration in the outlet was measured. At last, the gas flow bypassed the reactor again so as to verify the inlet concentration of Hg0. Meanwhile, a mercury conversion system coupled with RA-915 M mercury analyzer was employed to measure Hg0 and Hg2+ concentrations in outlet flue gas. As shown in Fig. 1, in the mercury conversion system, gas stream released from the reactor was divided into two streams, one for measuring total mercury (HgT) and the other for elemental mercury (Hg0). On one stream, an impinger with 10 wt% acidic stannous chloride (SnCl2) solution was used to measure HgT, since acidic SnCl2 solution can reduce Hg2+ to Hg0 which can then be detected by the mercury analyzer. On the other stream, an impinger containing 10 wt% potassium chloride (KCl) solution was used to measure Hg0, because KCl solution can capture Hg2+ and allow only Hg0 to pass through. Before the sampling gas entered the SnCl2 solution, a 10 wt% sodium hydroxide (NaOH) solution was used to remove SO2. The concentration of Hg2+ in outlet flue gas could then be calculated by the difference between HgT and Hg0. At the end of the conversion system, the two streams converged to a 10 wt% NaOH solution in which acid gases were captured to avoid corrosion of the detection cell in the mercury analyzer. Supposing mercury absorbed on AC was thoroughly in oxidation state. Therefore, the Hg0 removal efficiency (Eoxi) was quantified by the following equation:
Where [Hg0]in and [Hg0]out represent Hg0 concentration (μg/m3) at the inlet and outlet of the reactor, respectively. Since the mercury discharged from reactor includes both Hg0 and Hg2+, the Hg0 capture efficiency (Ecap) was introduced to be calculated by the formula below:
Where [Hg0]in represents Hg0 concentration (μg/m3) in inlet of the reactor, and [HgT]out represents HgT concentration (μg/m3) in outlet of the reactor.
Results and discussion
Samples characteristics
BET analysis
The physical properties of virgin AC and impregnated AC including BET surface area, pore volume, and average pore size are summarized in Table 2. The results showed that the modification increased BET surface area and pore volume. During impregnation and calcination, a certain number of new pores might be generated due to the reaction between AC and active components, which account for higher BET surface area of impregnated AC compared to virgin AC. However, the increasing doping of Fe2O3 reduced the BET surface area and pore volume of AC to some extent except for Fe4Ce2/AC. Especially, it can be observed that Fe3Ce3/AC had the lowest BET surface area of 276.74 m2/g and minimum pore volume of 0.14 cm3/g among impregnated AC. The result could be explained as follows: On one hand, it was possibly resulted from the deterioration of some of the thin pore walls on AC over strong oxidation of Fe-Ce mixed oxides. On the other hand, the internal pores might be blocked when Fe2O3 and CeO2 particles enter into the interior of pores (Wen et al. 2011).
XRD analysis
The XRD patterns of virgin AC, Ce6/AC, Fe2Ce4/AC, Fe3Ce3/AC, and Fe4Ce2/AC are presented in Fig. 2. Two diffraction peaks attributed to AC were detected at 26.66° and 44.58°. Nevertheless, the two peaks even disappeared when the mass ratio of Fe2O3 and CeO2 was above 2:4, indicating that Fe2O3 and CeO2 interacted with AC strongly in these samples. The peaks at 27.88°, 32.96°, 46.98°, and 56.14° were corresponding to CeO2, which could be detected over Ce6/AC. When the mass ratio of Fe2O3 and CeO2 was 2:4, 3:3, 4:2, there was no obvious peaks belong to CeO2 or Fe2O3. It may be explained that Fe2O3 and CeO2 existed as an amorphous phase or highly dispersed on the surface of AC. When the mass percentage of Fe single oxide was increased to 30 %, it could be seen that Fe mainly presented in the form of Fe2O3. Peaks with strong intensity appeared at 30.52°, 35.74°, 43.07°, and 62.68° which were matched well with peaks of Fe2O3. In addition, the peaks attributed to CeO2 had strong intensity for Fe15Ce15/AC. This implied that the synergy of Fe2O3 and CeO2 probably led to an excellent dispersion of active components on the surface of AC when the overall loading value was 6 %.

XRD spectra of virgin AC and the FexCey/AC samples
SEM analysis
Figure 3 shows SEM micrographs of virgin AC and the selected impregnated AC. The characteristics of AC surface have changed according to different active components loading. As shown in Fig. 3d, Fe2O3 and CeO2 were widely dispersed on the AC surface and only a few agglomerates existed in Fe3Ce3/AC. Furthermore, there were more small holes on the surface of Fe3Ce3/AC compared with other samples, which indicated that Fe2O3 and CeO2 extensively modified the structure of AC. However, in the case of Fe4Ce2/AC, the agglomerates became bigger. Consequently, there were synergistic effects between Fe2O3 and CeO2, so the interaction could improve the dispersion of Fe2O3 and CeO2 on the surface of AC and then improved the Hg0 removal efficiency. Moreover, the phenomenon was in accordance with XRD analysis. According to the refs (Huang et al. 2009; Sahoo et al. 2009; Wan et al. 2009), cerium can enhance the catalytic property owing to the MOx-CeOx (M = Fe, Cu and Pd) interaction. In our study, doped samples with layer of Fe2O3 and CeO2 covered on certain surface of the sorbents could provide enough oxidation capacity.

SEM micrographs of a virgin AC, b Ce6/AC, c Fe2Ce4/AC, d Fe3Ce3/AC, e Fe4Ce2/AC, f Fe6/AC
XPS analysis
To determine the chemical state and the relative portion of the main elements on the surface of different samples, fresh Fe3Ce3/AC and used Fe3Ce3/AC were investigated by XPS technique. The XPS spectra over the spectral regions of O 1 s, Fe 2p, Ce 3d, and Hg 4f are shown in Fig. 4.

O 1 s, Fe 2p, Ce 3d, and Hg 4f XPS spectra of fresh Fe3Ce3/AC and used Fe3Ce3/AC
In Fig. 4a, b, for fresh Fe3Ce3/AC, the O 1 s spectrum was divided into two main peaks. A binding energy of about 530.2 eV was ascribed to the lattice oxygen (denoted as Oα) in the metal oxides. The peak at higher binding energy (about 531.5 eV) belonged to chemisorbed oxygen and/or weakly bonded oxygen species (denoted as Oβ), which were regarded as the most active oxygen and played an important role in oxidation reaction (Gao et al. 2010; Kang et al. 2007; Li et al. 2011a, b). For used Fe3Ce3/AC, the peak with a binding energy of about 530.3 eV was attributed to the lattice oxygen (Liu et al. 2006), while the binding energy of 532.2 eV represented chemisorbed and/or weakly bonded oxygen (as in carbonyl or carboxyl groups) (Huang et al. 2005; Liu et al. 2006). The fitted peak of O 1 s in the vicinity of 532.6 eV corresponded to oxygen in hydroxyl and/or surface adsorbed water (denoted as Oγ) (Eom et al. 2008; Gao et al. 2010).
Hypothetically, the atomic concentration of total oxygen on AC was denoted by OT. After comparison of O 1 s profiles in fresh Fe3Ce3/AC and used Fe3Ce3/AC, the ratio of Oα/OT clearly decreased from 33.58 to 7.43 %. In this regard, it could be inferred that lattice oxygen took part in the reaction of removing Hg0. Besides, the value of Oβ/OT obviously decreased from 66.42 to 40.55 % after the adsorption and oxidation reaction. This clearly manifested that some chemisorbed oxygen and/or weakly bonded oxygen species were consumed in Hg0 removal.
The XPS spectra of Fe 2p on fresh Fe3Ce3/AC and used Fe3Ce3/AC are shown in Fig. 4c, d. Iron ions in FeO are characterized by the Fe2p3/2 binding energy in the range of 709.8–710.6 eV (Descostes et al. 2000; Graat and Somers 1996), so the peaks at 710.4 and 710.6 eV referred to Fe2+ cations. The binding energy concentrated on 711.6 eV was typical characteristic of Fe3+ (Xu et al. 007). After Hg0 was captured, the binding energy centered at about 712.1 eV which might be ascribed to Fe3+ cations bonded with hydroxyl groups(≡FeIII-OH). Furthermore, the peak at about 719 eV was the fingerprint of Fe3+ species (Shen et al. 2010). After Hg0 removal, a few changes concerning the proportion of valence happened.
Complicated XPS spectra of Ce 3d for fresh Fe3Ce3/AC and used Fe3Ce3/AC are presented in Fig. 4e, f. The peaks labeled u are due to 3d3/2 spin-orbit states, and those labeled v are the corresponding 3d5/2 states (Reddy et al. 2003). The u/v, u2/v2, and u3/v3 doublets represent the 3d104f0 state of Ce4+, while the doublet labeled u1/v1 represents the 3d104f1 initial electronic state corresponding to Ce3+ (Mullins et al. 1998). It was apparent that Ce4+ and Ce3+ were coexisting. The presence of Ce3+ could create charge imbalance, vacancies, and unsaturated chemical bonds on the surface of samples (Yang et al. 2006), which lead to the increase of surface chemisorbed oxygen. Based on the peak areas, Ce4+ is the predominant form and considered to be advantageous for Hg0 oxidation (Wan et al. 2011)). The ratio of Ce4+/Ce3+ decreased from 6.42 to 2.26 after removing Hg0, which manifested a reduction of Ce4+ during Hg0 oxidation reaction.
The XPS spectrum of Hg 4f for used Fe3Ce3/AC is shown in Fig. 4g. The binding energy centered at approximate 103.3 eV was attributed to Si 2p electron (Hua et al. 2010). The peak appeared at about 101.6 eV was assigned to HgO (Ji et al. 2008). However, no adsorbed Hg0 was found on the sample surface, which might be explained that the content of Hg0 was lower than the detection limit of XPS analysis or the adsorbed Hg0 desorbed from the surface of sample. Considering the results of O 1 s and Hg 4f, it could be certified that HgO was the product of oxygen-containing groups and Hg0, both of which were absorbed weakly on the sample surface.
Performance of the samples
Screening of the optimal sample
Figure 5 presents the effect of different mass ratio of Fe2O3 and CeO2 on Hg0 removal efficiency. In comparison to virgin AC, AC modified with Fe2O3 and CeO2, particularly when the mass ratio of Fe2O3 and CeO2 was 3:3, promoted the Hg0 removal significantly. Hg0 removal ability was enhanced with the increase of Fe2O3 when loading value was below 3 % and then slightly decreased with increasing Fe2O3 loading value to 4 %. Table 2 shows that Fe3Ce3/AC has the lowest BET surface area among impregnated AC. What’s more, Fe1Ce5/AC with the largest BET surface area has the minimum Hg0 removal efficiency of 73.89 % except for virgin AC. It indicated that Hg0 removal was mainly influenced by the oxidation reaction and less affected by BET surface area which could affect physical adsorption. Besides, the synergy between Fe2O3 and CeO2 should be a rational reason for the excellent performance of Fe3Ce3/AC. The aforementioned phenomenon was consistent with the result of SEM analysis. From the analysis above, it could be concluded that Hg0 removal was attributed to the combination of adsorption and oxidation. Furthermore, the lattice oxygen, chemisorbed oxygen, and/or weakly bonded oxygen species made a contribution to Hg0 oxidation. Specially, the reaction mechanism of Hg0 removal could be explicated as follows: gaseous Hg0 was firstly adsorbed on the sample surface to form Hg0(ad), then Hg0(ad) would react with the lattice oxygen which was released from Fe2O3 and CeO2 to form HgO(ad). The possible reactions can be described as follows:

Effect of the mass ratio of Fe2O3 and CeO2 on Hg0 removal efficiency. Reaction conditions: 70 μg/m3 Hg0, 5 % O2, 300 ppm NO, 400 ppm SO2, 12 % CO2, N2 as balance, GHSV = 4200 h−1, T = 110 °C
Furthermore, in order to explore the material balance among the amounts of Hg0 in the inlet and the outlet streams and the one captured by the adsorbent, a related experiment was carried out. In the experiment (the reaction condition was the same as Set 4 in Table 1), a mercury conversion system coupled with RA-915 M Mercury Analyzer was used to determine the concentration of Hg0 and Hg2+ in outlet flue gas. As shown in Table 3, it could be found that Eoxi and Ecap were nearly the same to each other, which indicated that the formed HgO was captured on the Fe3Ce3/AC. The result was in accordance with the analysis of XPS.
Screening of reaction temperature
A set of experiments were carried out to explore reaction temperatures (50–200 °C) on Hg0 removal. The results given in Fig. 6 showed that Hg0 removal efficiency increased with increasing the temperature from 50 to 110 °C and then decreased with the temperature rising from 110 to 200 °C. This phenomenon might be explained by the inhibition of physical adsorption of Hg0 at relatively high temperature. Therefore, a range of correspondingly low temperature was beneficial for the reaction between Hg0 and oxygen functional groups. So, the optimal temperature of 110 °C was chosen for further study.

Effect of reaction temperature on Hg0 removal efficiency. Reaction conditions: 70 μg/m3 Hg0, 5 % O2, 300 ppm NO, 400 ppm SO2, 12 % CO2, N2 as balance, GHSV = 4200 h−1, T = 50, 80, 110, 140, 170, 200 °C
To further investigate the durability of Hg0 removal over Fe3Ce3/AC in SFG, the breakthrough curve of Hg0 capture was carried out at 110 °C. As shown in Fig. 7, the C/C0 achieved an average value of 0.13 during the 14 h reaction, which implied excellent endurance with high activity of Fe3Ce3/AC.

The breakthrough curve of elemental mercury captured by Fe3Ce3/AC under simulated flue gas. Reaction conditions: 70 μg/m3 Hg0, 5 % O2, 300 ppm NO, 400 ppm SO2, 12 % CO2, N2 as balance, GHSV = 4200 h−1, T = 110 °C
Effects of individual flue gas components
Effect of O2
The Hg0 removal efficiency over Fe3Ce3/AC at 110 °C under pure N2 gas was observed to be around 46.00 %, as shown in Fig. 8. The loss of Hg0 in the absence of O2 could be ascribed to the consumption of lattice oxygen over the sample (He et al. 2014). When 5 % O2 was introduced to gas flow, the Hg0 removal efficiency increased to about 71.33 %. However, further increasing the O2 concentration to 8 %, the Hg0 removal efficiency did not increase any more. This result suggested that 5 % O2 would achieve the supplement for consumed lattice oxygen and was sufficient to sustain Hg0 oxidation.

Effect of individual flue gas components on Hg0 removal efficiency. Reaction conditions: GHSV = 4200 h−1, T = 110 °C
As illustrated in Fig. 9, the Fe3Ce3/AC has a relatively high activity in the presence of O2. When Hg0 vapor was introduced into (N2/5 % O2) mixture, O2 was found to have the capability of promoting Hg0 oxidation. Unlike the test with N2, the Hg0 removal efficiency was consistently much higher in (N2/5 % O2) mixture for the whole 12 h. Many researchers have put forward that active oxygen can be generated from adsorbed oxygen via cerium species (Deng et al. 2011; Wang et al. 2013). So O2 would provide metal oxides with oxygen to make sure that Hg0 oxidation could be sustained. Combined with the results of XPS, it indicated that the re-oxidization of formed Ce3+ and Fe2+ cations (reactions (7)–(8)) happened. Under the sufficient gas-phase O2 supply, some cation vacancies may be recovered. Reactions involved can be expressed as follows:

Breakthrough curves of elemental mercury captured by Fe3Ce3/AC under N2 and N2 plus 5 % O2. Reaction conditions: GHSV = 4200 h−1, T = 110 °C
Effect of NO
Compared with pure N2 gas, an addition of 300 ppm NO exhibited almost the same effect on Hg0 removal. With further increasing the concentration of NO, the Hg0 removal efficiency increased gradually. The Hg0 removal efficiency was promoted to approximate 74.64 % with respect to 800 ppm NO, which was even higher than that 5 % O2 balanced with N2 stream. The presence of Ce3+ on the sample surface was reported to promote the oxidation of NO-NO2, and NO2 was demonstrated to significantly improve heterogeneous oxidation of Hg0 over fly ash and activated carbon-based sorbents (Miller et al. 2000; Norton et al. 2003). Additionally, adding 5 % O2 into 300 ppm, NO increased the Hg0 removal efficiency to about 90.63 %. What interests us is that the addition of 5 % O2 coupled with 800 ppm NO into gas stream unexpectedly resulted in a distinct decrease of Hg0 removal efficiency. This phenomenon was in accordance with other researchers who hypothesized that HgO(ad) and NO might be generated between Hg0 and NO2 (Norton et al. 2003). The excess addition of NO tends to drive the equilibrium to the left which will inhibit Hg0 oxidation by this mechanism:
Effect of SO2
In literatures, the effect of SO2 on Hg0 removal in the flue gas is complicated and not conclusive. Promotional (Eswaran and Stenger 2005; Li et al. 2011a), inhibitive (Ji et al. 2008; Li et al. 2010), and negligible effects (Li et al. 2008) have been reported. As shown in Fig. 8, when the concentration of SO2 increased from 400 to 800 ppm, the Hg0 removal efficiency slightly decreased from 52.57 to 48.10 %. The addition of 1200 ppm SO2 could achieve an insignificant promotional effect on Hg0 removal, and the removal efficiency increased to around 69.00 % by further increasing the concentration to 2000 ppm. Adding 5 % O2 and 2000 ppm SO2 into the gas stream, the Hg0 removal was greatly enhanced as shown in Fig. 8. This result indicated that SO2 had a promotional effect on Hg0 removal with the aid of O2. In the presence of gaseous O2, abundant chemisorbed oxygen was generated due to Ce3+ related charge imbalance. SO2 was oxidized to SO3 by chemisorbed oxygen, which would generate new sites for Hg0 (Li et al. 2011b). Furthermore, SO3 could react with Hg0 to produce HgSO4 (Fan et al. 2010). The reactions are proposed as follows:
Effect of H2O(g)
H2O(g) is one of the main components in coal-fired flue gas and has inhibitive impacts on the activity of samples, so water resistance is an important parameter to assess for possible industrial application. H2O(g) has been reported to inhibit Hg0 oxidation over metal oxide catalysts due to competitive adsorption (Li et al. 2011a; Li et al. 2008). The same phenomenon was observed in our study. As listed in Fig. 8, the addition of 8 % H2O(g) into SFG caused 15.29 % declination of Hg0 removal efficiency. The inhibitory effect of H2O possibly came from its competitive occupancy of the available active sites and thus inhibited the Hg0 adsorption.
Conclusions
In the study, Fe2O3 and CeO2 modified AC was prepared and applied to remove Hg0 from simulated flue gas in a lab-scale fixed-bed system at 50–200 °C. Results showed that impregnated AC samples had higher capacity for Hg0 removal compared to virgin AC. Particularly, when the mass ratio of Fe2O3 and CeO2 was 3:3 and total mass percentage of Fe-Ce mixed oxides was 6 %, the Hg0 removal efficiency reached to an average of 88.29 % at 110 °C. Besides, the Hg0 removal efficiency was affected by flue gas components, therein, O2 and NO were beneficial for Hg0 removal. SO2 showed an insignificant inhibition for Hg0 removal, while it was observed to promote Hg0 removal in the presence of O2. In addition, the Hg0 removal efficiency had a slight declination when H2O (g) was added into the flue gas. The analysis of XPS indicated that the main species of mercury on used Fe3Ce3/AC was HgO, and the Hg0 oxidation benefited from lattice oxygen, chemisorbed oxygen, and/or weakly bonded oxygen species on the surface of Fe3Ce3/AC. Considering the necessity and cost savings, the following work should optimize the sample composition to investigate NOx and Hg0 removal simultaneously.
References
Ayastuy J, Gil-Rodríguez A, González-Marcos M, Gutiérrez-Ortiz M (2006) Effect of process variables on Pt/CeO2 catalyst behaviour for the PROX reaction. Int J Hydrogen Energ 31:2231–2242. doi:10.1016/j.ijhydene.2006.04.008
Cao Y, Chen B, Wu J et al (2007) Study of mercury oxidation by a selective catalytic reduction catalyst in a pilot-scale slipstream reactor at a utility boiler burning bituminous coal. Energ Fuel 21:145–156. doi:10.1021/ef0602426
Delgado J, Pérez-Omil J, Rodríguez-Izquierdo J, Cauqui M (2006) The role of the carbonaceous deposits in the catalytic wet oxidation (CWO) of phenol. Catal Commun 7:639–643. doi:10.1016/j.catcom.2006.02.003
Deng S, Liu H, Zhou W, Huang J, Yu G (2011) Mn–Ce oxide as a high-capacity adsorbent for fluoride removal from water. J Hazard Mater 186:360–1366
Descostes M, Mercier F, Thromat N, Beaucaire C, Gautier-Soyer M (2000) Use of XPS in the determination of chemical environment and oxidation state of iron and sulfur samples: constitution of a data basis in binding energies for Fe and S reference compounds and applications to the evidence of surface species of an oxidized pyrite in a carbonate medium. Appl Surf Sci 165:288–302. doi:10.1016/S0169-4332(00)00443-8
Dunham GE, DeWall RA, Senior CL (2003) Fixed-bed studies of the interactions between mercury and coal combustion fly ash. Fuel Process Technol 82:197–213. doi:10.1016/S0378-3820(03)00070-5
Eom Y, Jeon SH, Ngo TA, Kim J, Lee TG (2008) Heterogeneous mercury reaction on a selective catalytic reduction (SCR) catalyst. Catal Lett 121:219–225. doi:10.1007/s10562-007-9317-0
Eswaran S, Stenger HG (2005) Understanding mercury conversion in selective catalytic reduction (SCR) catalysts. Energ Fuel 19:2328–2334. doi:10.1021/ef050087f
Fan X, Li C, Zeng G et al (2010) Removal of gas-phase element mercury by activated carbon fiber impregnated with CeO2. Energ Fuel 24:4250–4254. doi:10.1021/ef100377f
Fan X, Li C, Zeng G et al (2012) Hg0 removal from simulated flue gas over CeO2/HZSM-5. Energ Fuel 26:2082–2089. doi:10.1021/ef201739p
Galbreath KC, Zygarlicke CJ (2000) Mercury transformations in coal combustion flue gas. Fuel Process Technol 65:289––10. doi:10.1016/S0378-3820(99)00102-2
Gao X, Jiang Y, Zhong Y, Luo Z, Cen K (2010) The activity and characterization of CeO2-TiO2 catalysts prepared by the sol–gel method for selective catalytic reduction of NO with NH3. J Hazard Mater 74:34––39. doi:10.1016/j.jhazmat.2009.09.112
Gillot B, Laarj M, Kacim S (1997) Reactivity towards oxygen and cation distribution of manganese iron spinel Mn3-xFexO4 (0 ≤ x ≤ 3) fine powders studied by thermogravimetry and IR spectroscopy. J Mater Chem 7:827–831. doi:10.1039/A607179A
Graat PC, Somers MA (1996) Simultaneous determination of composition and thickness of thin iron-oxide films from XPS Fe 2p spectra. Appl Surf Sci 100:36–40. doi:10.1016/0169-4332(96)00252-8
He C, Shen B, Chen J, Cai J (2014) Adsorption and oxidation of elemental mercury over Ce-MnOx/Ti-PILCs. Environ Sci Technol 48:7891–7898. doi:10.1021/es5007719
Hsi HC, Lee HH, Hwang JF, Chen W (2010) Mercury speciation and distribution in a 660-megawatt utility boiler in Taiwan firing bituminous coals. J Air Waste Manage Assoc 60:514–522. doi:10.3155/1047-3289.60.5.514
Hua X, Zhou J, Li Q, Luo Z, Cen K (2010) Gas-phase elemental mercury removal by CeO2 impregnated activated coke. Energ Fuel 24:5426–5431. doi:10.1021/ef100554t
Huang G, Liaw BJ, Jhang CJ, Chen YZ (2009) Steam reforming of methanol over CuO/ZnO/CeO2/ZrO2/Al2O3 catalysts. Appl Catal A: Gen 358:7–12
Huang JY, Shao YX, Huang HG et al (2005) Binding mechanisms of methacrylic acid and methyl methacrylate on Si (111)-7 × 7 effect of substitution groups. J Phys Chem B 109:19831–19838. doi:10.1021/jp0531659
Itaya Y, Kawahara K, Lee CW (2009) Dry gas cleaning process by adsorption of H2S into activated cokes in gasification of carbon resources. Fuel 8:1665–1672. doi:10.1016/j.fuel.2009.04.005
Jastrząb K (2012) Properties of activated cokes used for flue gas treatment in industrial waste incineration plants. Fuel Process Technol 101:16–22. doi:10.1016/j.fuproc.2011.05.028
Ji L, Sreekanth PM, Smirniotis PG, Thiel SW, Pinto NG (2008) Manganese oxide/titania materials for removal of NOx and elemental mercury from flue gas. Energ Fuel 22:2299–2306. doi:10.1021/ef700533q
Kamata H, Ueno SI, Sato N, Naito T (2009) Mercury oxidation by hydrochloric acid over TiO2 supported metal oxide catalysts in coal combustion flue gas. Fuel Process Technol 90:947–951. doi:10.1016/j.fuproc.2009.04.010
Kang M, Park ED, Kim JM, Yie JE (2007) Manganese oxide catalysts for NOx reduction with NH3 at low temperatures. Appl Catal A: Gen 327:61–269. doi:10.1016/j.apcata.2007.05.024
Kaspar J, Fornasiero P, Graziani M (1999) Use of CeO2-based oxides in the three-way catalysis. Catal Today 50:285–298. doi:10.1016/S0920-5861(98)00510-0
Li H, Li Y, Wu CY, Zhang J (2011a) Oxidation and capture of elemental mercury over SiO2-TiO2-V2O5 catalysts in simulated low-rank coal combustion flue gas. Chem Eng J 69:86–93. doi:10.1016/j.cej.2011.03.003
Li HL, Wu CY, Li Y, Zhang JY (2011b) CeO2–TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas. Environ Sci Technol 45:7394–7400. doi:10.1021/es2007808
Li J, Yan N, Qu Z et al (2010) Catalytic oxidation of elemental mercury over the modified catalyst Mn/α-Al2O3 at lower temperatures. Environ Sci Technol 44:426–431. doi:10.1021/es9021206
Li Y, Lee C, Gullett B (2003) Importance of activated carbon's oxygen surface functional groups on elemental mercury adsorption. Fuel 82:451–457. doi:10.1016/S0016-2361(02)00307-1
Li Y, Murphy PD, Wu CY, Powers KW, Bonzongo JCJ (2008) Development of silica/vanadia/titania catalysts for removal of elemental mercury from coal-combustion flue gas. Environ Sci Technol 42:5304–5309. doi:10.1021/es8000272
Li Z, Wu L, Liu H, Lan H, Qu J (2013) Improvement of aqueous mercury adsorption on activated coke by thiol-functionalization. Chem Eng J 228:925–934. doi:10.1016/j.cej.2013.05.063
Liu TY, Liao HC, Lin CC, Hu SH, Chen SY (2006) Biofunctional ZnO nanorod arrays grown on flexible substrates. Langmuir 22:5804–5809. doi:10.1021/la052363o
Milford JB, Pienciak A (2009) After the clean air mercury rule: prospects for reducing mercury emissions from coal-fired power plants. Environ Sci Technol 43:2669–2673. doi:10.1021/es802649u
Miller SJ, Dunham GE, Olson ES, Brown TD (2000) Flue gas effects on a carbon-based mercury sorbent. Fuel Process Technol 65:343–363. doi:10.1016/S0378-3820(99)00103-4
Mullins D, Overbury S, Huntley D (1998) Electron spectroscopy of single crystal and polycrystalline cerium oxide surfaces. Surf Sci 409:307–319. doi:10.1016/S0039-6028(98)00257-X
Norton GA, Yang H, Brown RC, Laudal DL, Dunham GE, Erjavec J (2003) Heterogeneous oxidation of mercury in simulated post combustion conditions. Fuel 82:107–116. doi:10.1016/S0016-2361(02)00254-5
Ogriseck S, Vanegas GPG (2010) Experimental investigations of ammonia adsorption and nitric oxide reduction on activated coke. Chem Eng J 160:641–650. doi:10.1016/j.cej.2010.04.004
Pavlish JH, Sondreal EA, Mann MD (2003) Status review of mercury control options for coal-fired power plants. Fuel Process Technol 82:89–165. doi:10.1016/S0378-3820(03)00059-6
Pitoniak E, Wu CY, Mazyck DW, Powers KW, Sigmund W (2005) Adsorption enhancement mechanisms of silica-titania nanocomposites for elemental mercury vapor removal. Environ Sci Technol 39:1269–1274. doi:10.1021/es049202b
Presto AA, Granite EJ (2006) Survey of catalysts for oxidation of mercury in flue gas. Environ Sci Technol 40:5601–5609. doi:10.1021/es060504i
Reddy BM, Khan A, Yamada Y, Kobayashi T, Loridant S, Volta JC (2003) Structural characterization of CeO2-TiO2 and V2O5/CeO2-TiO2 catalysts by Raman and XPS techniques. J Phys Chem B 107:5162–5167. doi:10.1021/jp0344601
Sahoo S, Mohapatra M, Pandey B, Verma H, Das R, Anand S (2009) Preparation and characterization of α-Fe2O3–CeO2 composite. Mater Charact 60:25–431. doi:10.1016/j.matchar.2008.11.006
Sakanishi K, Wu Z, Matsumura A (2005) Simultaneous removal of H2S and COS using activated carbons and their supported catalysts. Catal Today 104:94–100. doi:10.1016/j.cattod.2005.03.060
Shawwa AR, Smith DW, Sego DC (2001) Color and chlorinated organics removal from pulp mills wastewater using activated petroleum coke. Water Res 35:745–749. doi:10.1016/S0043-1354(00)00322-5
Shen B, Liu T, Zhao N, Yang X, Deng L (2010) Iron-doped Mn-Ce/TiO2 catalyst for low temperature selective catalytic reduction of NO with NH3. J Environ Sci 22:1447–1454. doi:10.1016/S1001-0742(09)60274-6
Wan H, Li D, Dai Y (2009) Effect of CO pretreatment on the performance of CuO/CeO2/γ-Al2O3 catalysts in CO + O2 reactions. Appl Catal A: Gen 60:26–2. doi:10.1016/j.apcata.2009.02.046
Wan Q, Duan L, He K, Li J (2011) Removal of gaseous elemental mercury over a CeO2-WO3/TiO2 nanocomposite in simulated coal-fired flue gas. Chem Eng J 170:512–517. doi:10.1016/j.cej.2010.11.060
Wang X, Zheng Y, Lin J (2013) Highly dispersed Mn–Ce mixed oxides supported on carbon nanotubes for low-temperature NO reduction with NH3. Catal Commun 7:96–99. doi:10.1016/j.catcom.2013.03.035
Wen X, Li C, Fan X et al (2011) Experimental study of gaseous elemental mercury removal with CeO2/γ-Al2O3. Energ Fuel 25:2939–2944. doi:10.1021/ef200144j
Xie Y, Li C, Zhao L (2015) Experimental study on Hg0 removal from flue gas over columnar MnOx-CeO2/activated coke. Appl Surf Sci 333:59–67. doi:10.1016/j.apsusc.2015.01.234
Xu Z, Zhao L, Pang F, Wang L, Niu C (2007) Partial oxidation of methane to synthesis gas over hexaaluminates LaMAl11O19-δ catalysts. J Nat Gas Chem 6:0–63. doi:10.1016/S1003-9953(07)60027-9
Yan N, Chen W, Chen J et al (2011) Significance of RuO2 modified SCR catalyst for elemental mercury oxidation in coal-fired flue gas. Environ Sci Technol 45:5725–5730. doi:10.1021/es200223x
Yang S, Zhu W, Jiang Z, Chen Z, Wang J (2006) The surface properties and the activities in catalytic wet air oxidation over CeO2-TiO2 catalysts. Appl Surf Sci 25:8499–8505. doi:10.1016/j.apsusc.2005.11.067
Yue L, Zhang XM (2009) Structural characterization and photocatalytic behaviors of doped CeO2 nanoparticles. J Alloy Compd 475:702–705. doi:10.1016/j.jallcom.2008.07.096
Zhang A, Zheng W, Song J, Hu S, Liu Z, Xiang J (2014) Cobalt manganese oxides modified titania catalysts for oxidation of elemental mercury at low flue gas temperature. Chem Eng J 236:29–38. doi:10.1016/j.cej.2013.09.060
Acknowledgments
The project is supported by the National Natural Science Foundation of China (51278177, 51478173) and the National High Technology Research and Development Program of China (863 Program, No. 2011AA060803).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article

Cite this article
Wang, Y., Li, C., Zhao, L. et al. Study on the removal of elemental mercury from simulated flue gas by Fe2O3-CeO2/AC at low temperature. Environ Sci Pollut Res 23, 5099–5110 (2016). https://doi.org/10.1007/s11356-015-5717-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-5717-7