
Abstract
The commonly observed enrichment of middle rare earth elements (MREE) in water sampled in acid mine drainage (AMD)-impacted areas was found to be the result of preferential release from the widespread mineral pyrite (FeS2). Three different mining-impacted sites in Europe were sampled for water, and various pyrite samples were used in batch experiments with diluted sulphuric acid simulating AMD-impacted water with high sulphate concentration and high acidity. All water samples independent on their origin from groundwater, creek water or lake water as well as on the surrounding rock types showed MREE enrichment. Also the pyrite samples showed MREE enrichment in the respective acidic leachate but not always in their total contents indicating a process-controlled release. It is discussed that most probably complexation to sulphite (SO3 2−) or another intermediate S-species during pyrite oxidation is the reason for the MREE enrichment in the normalized REE patterns.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rare earth elements (REE) are a group of elements with a high potential as process indicators in the system rock or soil and water. They are a group of f-block metals constituting the elements of La through Lu, being often subdivided into light REE (LREE; La to Pm), middle REE (MREE; Sm to Dy) and heavy REE (HREE; Ho to Lu). In the last years, they have become of increasing importance in environmental studies because of their release from a wide range of industrial processes. Furthermore, a more detailed understanding of REE cycles in the bio-geo-system is needed in times of their increasing need in green technologies and times of securing worldwide supply. Hence, it gets more and more important to study REE patterns, their natural background and REE’s geochemical behaviour in the system soil/rock and water. A REE pattern is the plot of concentrations normalized to a suitable standard (here: Post Archean Australian Shale = PAAS; McLennan 1989). This normalization is important in terms of interpretation since elements with even atomic number have a higher abundance than those with an odd one and thus, without normalization the graph along the series of REE would occur as zigzag pattern.
Acid mine drainage (AMD)-impacted areas often are featured by an enrichment in REE either in solution or in solids as summarized by Grawunder and Merten (2012). A very abundant phenomenon in AMD-impacted but also naturally acidic areas are normalized concave-shaped REE patterns in water indicating MREE enrichment, which has been widely discussed by various workers (e.g. Gimeno Serrano et al. 2000; García et al. 2007; Grawunder et al. 2010; Johannesson and Lyons 1995; Johannesson and Zhou 1997; Merten et al. 2005; Pérez-López et al. 2010; Welch et al. 2009). Previous studies excluded organic colloids, REE phosphate complexes (Johannesson et al. 1996) and SO4 2− complexation (Johannesson and Zhou 1999) to explain MREE enrichment and suggested the dissolution of primary and secondary minerals as a source. Amongst others, sulphate efflorescent salts and poorly crystalline iron oxyhydroxysulphates (Pérez-López et al. 2010), secondary Fe-Mn oxides/oxyhydroxides (Johannesson and Zhou 1999) or phosphatic minerals such as apatite containing MREE enrichment (Hannigan and Sholkovitz 2001) have been proposed. Especially apatite and uraninite were found to be featured by MREE enrichment when referring to the total contents (Grandjean-Lécuyer et al. 1993; Lécuyer et al. 2004; McLennan 1994; Reynard et al. 1999). Further, it was maintained that the relative total content REE patterns of a mineral species are almost similar due to the concept of “crystallographic/mineralogical control” (Bau and Möller 1992; Morgan and Wandless 1980). Besides these numerous approaches, literature shows that water having MREE enrichment is varying in terms of host rocks from Silurian slates to volcanic rocks to lignites (Bozau et al. 2004; Elderfield et al. 1990; Johannesson and Zhou 1999; Merten et al. 2005; Smedley 1991). To summarize up, there remains uncertainty over specific sources and processes causing widespread MREE enrichment in AMD-impacted, but also naturally acidic areas. Anyway, there are two main things nearly all AMD-impacted areas have in common: (1) low pH together with (2) the occurrence of sulphides like pyrite (FeS2), which is probably the most widespread sulphide mineral on Earth. As far as known, the REE release from pyrite was never taken into account, probably under the assumption that the release pattern is similar to the total content pattern. A previous work by Grawunder (2010) presented some first data on REE release from pyrite, which are also included in this work for further considerations. However, for the recent work only the MREE enrichment in mining-impacted sites with acidic pH will be taken into account. Buffering reactions leading to higher or neutral pH such as for example in the Yorkshire Pennine Orefield (Jones et al. 2013) would probably already have caused fractionations among the REE, since REE do not longer behave conservatively at higher pH (e.g. Verplanck et al. 2004). Furthermore, for the recent work it is expected that REE are, when talking about the solid phase, not only contained in REE minerals like monazite, but rather in all abundant minerals of a study site even though in lower content. As a first step of downscaling REE release in acidic areas to the mineral scale this work will especially focus on pyrite (FeS2). Based on three different mining-impacted areas, it is shown that MREE enrichment occurs independently on geological background and water system (groundwater, river and lake). Consequently, the idea of this study was to focus on the release of REE from pyrite under acidic conditions to assess whether pyrite is a possible and likely source of the widely discussed phenomenon of MREE enrichment. A batch approach under acidic and high sulphate conditions and therefore mimicking AMD impact provides evidence that REE patterns released from pyrite under these conditions are not equal to the total content REE patterns in pyrite.
The field sites
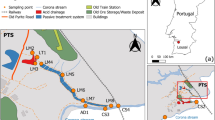
Water was sampled from three mining-impacted field sites within Europe in order to check if there MREE enrichment is also a present feature. From two of them also pyrite could be sampled (Germany and Romania). To increase the size of the database, additional pyrite material was provided by the Mineralogical Collection of the University of Jena and from private collections.
Ronneburg, Germany (GTF)
The German site is a part of the former Uranium mining area Ronneburg, East Thuringia. The sampling site, test site “Gessenwiese”, was created at the remediated basement area of the former leaching heap of Gessen. There, until 1989, low-graded ore, which was mainly Silurian and Ordovician shale, was leached for Uranium with acid mine drainage and sulphuric acid (10 g L−1) (Wismut GmbH 1994). Below the sealing of the heap Quaternary sediments of a limno-fluvial environment can be found (Grawunder et al. 2009). After remediation, in 2004 the test site Gessenwiese was installed in this area aiming at the improvement of phytoremediation strategies. Previous investigations found that the REE in the sediments were not strongly enriched or depleted compared to PAAS standard (Grawunder et al. 2009). However, REE concentrations in groundwater were very high with a total concentration of up to 8.15 mg L−1 (Grawunder and Merten 2012). Samples for this work were taken in April 2012 from five groundwater measuring points. Additionally, at this site a piece of shale with spherical pyrite of about 1 cm in diameter was sampled (samples 1a and 1b).
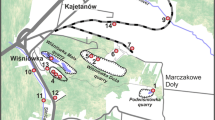
Haneş, Romania (RZ)
Haneş belongs to a big ore field close to Zlatna, where a vein of the Larga deposit, which is rich in Cu, Zn, Pb and Ag, was mined until the mid-1990s (Cook and Ciobanu 2004; Cook et al. 2009). Furthermore, pyrite lenses and the occurrence of sphalerite (ZnS) are described (Cook and Ciobanu 2004). Generally, ore deposits in the South Apuseni Mountains are associated with volcanic and subvolcanic rocks (Neubauer et al. 2005). During the screening in 2010, the small creek collecting the water from the Haneş mine (creek Almăşel, Duma 2009; RZ4, 5, 6) and a mine adit (RZ2) from Mina Haneş were sampled. At this site, two pyrite samples were taken (samples 11a and 11b).
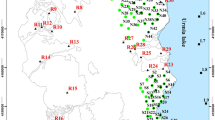
Kvarntorp, Sweden (SP)
A small acidic non-stratified lake, Lake Pölen (Karlsson et al. 2012), was sampled in the Kvarntorp district, about 20 km in the south of Örebro, Sweden. It belongs to a series of former open pits where Cambrian alum shale was mined and processed for the recovery of hydrocarbons (Karlsson et al. 2012). This shale is described in literature to contain amongst others quartz, micas (muscovite, biotite and chlorite), calcite veins as well as thick lenses of pyrite, which also occurs in microscopic and submicroscopic crystals (Assarsson and Grundulis 1961). Indeed, this acidic lake receives water from groundwater and shale residues (Karlsson et al. 2012), and not directly from a mine such as the Romanian site. Water samples were taken from the lake in June 2011 from 1 and 2.5 m depth (SP1 and SP2, respectively). No pyrite could be sampled.
Materials and methods
Equipment
Clean bottles used for water sampling in field were rinsed twice with sample. For the preparation of the chemicals applied in the following ultrapure water (0.055 μS cm−1; Purelab Plus, USF Seral) was used. The used pH measuring unit (pH meter + pH electrode) was calibrated at each measuring day using pH 4.01 and pH 7.00 standards (WTW).
Water sampling and analytics
Physicochemical parameters of the water samples were measured in situ using a portable instrument Multi 340i (WTW) with TetraCon 325 (Electrical Conductivity, EC), Schott BlueLine 31 Rx (3 M KCl; Eh), WTW SenTix 81 (3 M KCl, pH and temperature). Eh was corrected to temperature and rounded to the nearest tens. Samples for anions (except for HCO3 −) and elements were filtered using glass fibre prefilters (Sartorius) and cellulose acetate filters (0.45 μm, Sartorius). Water samples for element analysis finally were acidified with HNO3 (65 %, subboiled) to pH < 2 for stabilization. All samples were kept at 6 °C until analysis.
HCO3 − was determined by titration of total alkalinity (Titrino 716 DM, Metrohm), whereas Cl−, F− and SO4 2− were measured by means of ion chromatography (DX-120, Dionex). For water samples, digestion solutions and leaching solutions, the elements Al (for GTF samples and total digestions), Ca, Fe, K, Mg, Mn, Na, Zn (only for RZ samples), as well as S only for elution solutions were determined using inductively coupled plasma-optical emission spectrometry (ICP-OES; Varian 725 ES). Inductively coupled plasma-mass spectrometry (ICP-MS; X-Series II, ThermoFisher Scientific) was used for analysing the elements Al (for all other samples), As, Cd, Co, Cu, Ni, Pb, Zn and REE (La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu). Solutions were measured three times and tested for outliers by the method of Grubb (for N = 3 and P = 90 %). Given values are mean values.
Samples for dissolved organic carbon (DOC) were filtered in field to 0.45 μm (glass fibre prefilters, Sartorius; cellulose acetate filters, Sartorius) into 40 mL glass bottles and analysed later by using a TOC Analyser (multi N/C 2100, Analytik, Jena).
All diagrams have been created with either Origin 8.5.0G (OriginLab Corporation) or MS Excel (2010) and graphically formatted with CorelDraw X5 (Corel Corporation).
Mineralogy, geochemistry and leaching
Solid samples were dried at 35 to 40 °C and ground subsequently depending on the amount either by using a centrifugal ball mill for low amounts (MM400, grinding jar and ball zirconium oxide, Retsch) or a vibration grinding mill for higher amounts (HSM 100A, agate grinding vessels, Herzog). Only for samples with strongly weathered surface (1a and 1b sampled at the German site; Table 1) 35 % HCl (Suprapur, Roth) was used to clean the surfaces before grinding.
X-ray diffractometry (XRD) of all samples was carried out using a Bruker D8 Advance DaVinci diffractometer with Cu kα radiation, 40 kV and 40 mA, a step size of 0.02° 2θ within the range of 5–80° 2θ and a measuring time of 0.5 s. Mineral phases were identified using the software package Diffrac.suite EVA v1.4 (Bruker AXS, with database ICDD PDF-2 release 2011). The identified samples were proven to be pyrite or in three cases to contain pyrite within a polymineral sample (samples 7, 10 and 17; Table 1).
For total digestion, 100–150 mg of each ground sample were digested in a pressure digestion system (DAS, PicoTrace). Therefore, the ground material was put in TFM vessels and 2 mL 65 % HNO3 (subboiled), 3 mL 40 % HF and 3 mL 70 % HClO4 (both Suprapur, Merck) were added. This mixture was then heated up to 180 °C within 6 h. After maintaining the temperature for 12 h the samples were cooled down. For evaporating the acids, the system was heated up again to 180 °C for 4–5 h in a special evaporating hood maintaining the temperature again for 12 h. The remaining solids were dissolved again after adding 2 mL HNO3 (subboiled), 0.6 mL HCl (Suprapur, Roth) and 7 mL ultrapure water (Purelab Plus, USF Seral) at 150 °C within 8 h. The cooled samples were transferred to calibrated 25 mL flasks (PMP, Vitlab) and replenished to 25 mL by addition of ultrapure water to determine the total contents of Al, As, Ca, Co, Cu, Fe, Mg, Mn, Ni, Pb, Zn and REE by ICP-OES and ICP-MS as described above. This method is well suited for measuring of metals, but S, which is of special relevance when working with pyrite, can partially escape from the vessel because of its volatile behaviour. In the following, results from bulk chemistry analysis are labelled with “TY…”. Digestion results for samples 1–5 were taken from Grawunder (2010).
To obtain the release of REE from the pyrite and polymineral samples, they were ground as described above to get a comparable grain size. This powder was leached with 10 g L−1 H2SO4 (made of H2SO4, 95–97 %, p.A., Merck) in 50 mL tubes (Greiner bio-one) for 24 h by shaking end-over-end (ELU, E. Bühler) in triplicates. Additional blanks (just acidic solution) were prepared the same way. H2SO4 in this concentration was chosen for batch leaching for the following reasons: (1) acidic conditions and high SO4 2− concentrations are typical for AMD-impacted areas; (2) at the first sampled site (Germany), 10 g L−1 H2SO4 were formerly used for heap leaching (Wismut GmbH 1994). The amount of solid material used in the leachings was ranging between about 1 to 5 g, depending on the available sample amount. The liquid/solid ratio during the experiments was 10:1 (10 mL per 1 g). Results for leachings of Y1 to Y5 at a liquid/solid ratio of 9:1 have been taken from Grawunder (2010). After elution, samples were centrifuged (15 min at 3,000 rpm; Multifuge 3L, Heraeus), then the solution was filtered to 0.45 μm (Y1-5: cellulose acetate filters, Sartorius; all others: GHP acrodisc, PALL), acidified with one drop HNO3 (65 %, subboiled) for stabilization and stored cool until analysis by ICP-MS and ICP-OES as described above. Additionally, directly after 24 h of leaching, pH was measured (WTW pH 330 with WTW SenTix 81; 3 M KCl) in the supernatant of the elutions. In the following, leaching results are labelled with “Y…”.
Calculations estimating the dissolved amount of pyrite were carried out based on bulk Fe and dissolved Fe content under the assumption that the samples are almost exclusively pure pyrite. Hence, for these calculations only pyrite samples were taken into account not being part of a polymineral sample as previously identified with XRD (compare Table 1). [S]sol representing the sulphur from pyrite dissolved in solution was calculated by subtraction of the blank, which is only the acid, from the concentration measured in the elution solution after 24 h of batch experiment. [Fe]sol was measured by ICP-OES, as described above, representing the dissolved Fe. Both were used as molar concentration to calculate the factor R = [S]sol/[Fe]sol according to Ichikuni (1960) and Descostes et al. (2004). When the R value is 2, the dissolution is stoichiometric. Lower values indicate non-stoichiometric dissolution, which is the expected range under acidic conditions (Ichikuni 1960; Descostes et al. 2004). Only pyrite samples being not part of a polymineral sample were taken into consideration for calculations of R (compare Table 1).
Rare earth elements: ratios and anomalies, evaluation of MREE enrichment
Among the REE, Ce, Eu and Gd can occur with either positive or negative anomalies what is mainly due to different geochemical behaviour or anthropogenic influence. For the calculation of Ce and Eu anomalies the following Eqs. 1 and 2 (Bau and Dulski 1996) are applied using PAAS (McLennan 1989) normalized values indexed with “PAAS”. Resulting values <0.8 indicate a negative anomaly, values >1.2 a positive one following the standard deviations calculated for the Eu anomalies in the leaching solutions.
For comparison of the subgroups of light, middle and heavy REE, ratios of PAAS-normalized representatives, that means La for LREE, Tb for MREE and Lu for HREE, are calculated ([Tb/La]PAAS, [Lu/La]PAAS and [Lu/Tb]PAAS). Values higher than 1.2 indicate enrichment; values below 0.8 indicate depletion of a subgroup relative to another one. This classification follows the standard deviations calculated for the ratios in the leaching.
In aqueous samples, MREE enrichment can be overlain by LREE or HREE enrichment, e.g., due to mixing of waters as demonstrated for two samples A and B at different mixing ratios in Fig. 1. Of course, such a “mixed pattern” occurs not only after mixing of two waters, but can also occur during release of different REE patterns from different minerals that occur together in a host rock. Thus, the release pattern of a rock sample finally represents the mixed release patterns of different single minerals. For this model, under acidic conditions strong fractionations e.g. by formation of secondary minerals are neglected, since REE behave conservatively in acidic sulphate waters as e.g. reported by Verplanck et al. (2004) for water with pH below 5.1. Figure 1 is only to clarify that the calculation of REE ratios as well as the optical impression of a REE pattern can lead to a non-consideration of a present MREE enrichment during release due to its weaker appearance.

Sketch illustrating the change in the REE pattern at different mixing ratios of two aqueous samples A and B. Assuming that A and B are mixed according to the different mixing ratios, the resulting REE pattern will have a stronger or weaker pronounced MREE enrichment, which especially for release from polymineral samples in this study could be of importance. For this model REE are considered to behave conservatively
Hence, considering mixing of different REE release patterns especially for minerals within the polymineral samples, as “MREE-enriched patterns”, patterns will be termed that have either a [Tb/La]PAAS ratio > 1.2, a [Lu/Tb]PAAS ratio <0.8, or both of them. This includes also the effect of pattern mixing due to sample impurities by other mineral species.
Statistical analysis
To check if the intensity of MREE enrichment is related to the release of other elements from pyrite, a principle component analysis (PCA) with Varimax rotation was carried out using SPSS 19.0.0 (IBM). Therefore, released contents of REE (La-Lu), As, Al, Ca, Co, Fe, Mg, Mn, Ni, Pb and Zn were implemented to the software. Eu was excluded due to its anomalous behaviour. Cu was excluded because of four values being below detection limit. Further the samples 7, 10 and 17 were excluded, since all of them contain mineral impurities. Also sample 1b was excluded due to its high similarity to 1a. The MREE enrichment was taken into account as a variable calculated with PAAS-normalized values for La, Tb and Lu in a similar way than the anomalies.
Results
Water samples: general hydrochemistry
All water samples show an acidic pH in the range of 2.9 to 5.6, and EC values between 1.3 and 13.9 mS cm−1 (Table 2). The Eh values indicate oxidizing conditions (450–750 mV) for all the samples in this study. The dominating anion in all water samples is, as expected for mining-impacted environments, SO4 2−. All waters have as dominating cation Mg, Ca or both of them (referred to meq L−1 concentrations). For samples RZ2 and RZ4 quite high Fe concentrations of 0.54 and 1.50 g L−1, respectively, have been measured.
Further, Al is an important metal for all sites with concentrations attaining values up to 237 mg L−1. Ni occurs especially in the German site in high concentrations up to 55 mg L−1 (Table 2). Extreme values of Zn were analysed at the Romanian site (up to 193 mg L−1). The DOC is in the range of 0.3 to 7.6 mg L−1.
Water samples: rare earth elements
In this study, the maximum total REE concentration was measured for GTF41 (4.7 mg L−1; Table 2). The PAAS-normalized REE patterns of all working areas show a typical MREE enrichment ([Tb/La]PAAS-range = 4.8–19.1; [Lu/Tb]PAAS-range = 0.5–0.9; Fig. 2; Table 2). The occurrence of anomalies differs from site to site. Samples from the German site show a positive Ce anomaly (Ce/Ce* = 1.2–3.5) and most of the the Romanian samples a positive Eu anomaly (Eu/Eu* = 1.1–1.6; Table 2).

PAAS-normalized REE patterns of water sampled at the different mining sites. All of them show a typical clear MREE enrichment (highlighted with grey background), partially overlain by a HREE enrichment. Furthermore, German samples show a distinct positive Ce anomaly, some Romanian samples a positive Eu anomaly. The two Swedish samples are almost identical and overlay each other. GTF Germany, RZ Romania, SP Sweden
Total contents and leaching of the sulphides
Besides Fe (323 to 472 mg g−1) and REE, for all samples chemical impurities have been taken into account (Table 3). For the pyrite samples especially Al (up to 11.4 mg g−1), As (up to 2.8 mg g−1), Ca (up to 5.5 mg g−1), but also Co, Cu, Mg, Mn, Ni, Pb and Zn were found in high total contents. For the polymineral samples, Cu was with 13.6 and 13.5 mg g−1, respectively, especially high in TY7 and TY10. Furthermore, for the polymineral samples TY7, TY10 and TY17 also high contents of Ca (3.4–26.2 mg g−1) and Mg (5.8–42 mg g−1) were measured underlining the presence of other minerals such as dolomite or calcite (compare Table 1).
The released element contents from the pyrite samples (Table 3) are especially high for Fe (1.5–9.8 mg g−1) and Ca (0.8–6.1 mg g−1), as well as Al (0.02–0.83 mg g−1), Mg (0.01–0.97 mg g−1), Mn (3.5–712 μg g−1) and partly Zn (0.7–285 μg g−1). For Mg (up to 10 mg g−1) and Zn (up to 1,719 μg g−1) again the polymineral samples Y7, Y10 and Y17 exceed these values. Furthermore, it is remarkable that Ca for samples 1a, 1b, 5, 8, 10, 12, 13 and 14 as well as Mn for sample 5 is in the leachate in a range of 100 ± 10 % of the total content indicating complete dissolution. Al, As, Co, (Cu), Fe and Ni release is with ≤33 % comparably low (Table 3).
The total REE contents of the pyrites were in the range of about 1.5 in TY9 to 66.3 μg g−1 in TY3 (Table 1). The PAAS-normalized total content REE patterns are slightly different for all samples (Fig. 3). Some are enriched in HREE (e.g. TY3, TY4, TY10, TY12, TY13, TY14 and TY15), others in MREE (e.g. TY1a and b, TY7 and TY8) and yet others have positive Eu anomalies (TY5, TY7, TY12 and TY14) or slightly negative ones (TY3). The Romanian pyrite samples (11a and 11b) will not be taken into account since most REE in total and leached contents were below detection limit. No statement on MREE enrichment is possible for those samples.

PAAS-normalized REE patterns of total (black lines, TY…) and leached contents (grey lines, Y…) from pyrite and polymineral samples containing pyrite (compare Table 1). All samples show a MREE enrichment in the REE pattern of the leaching solution. Please note different scaling of y-axes for samples 12, 13 and 14. Data for samples 1a, 1b, 3, 4 and 5 are taken from Grawunder (2010)
The pH in the leaching solution of the samples (Table 4) did change only up to 0.2 pH units compared to the initial elution agent (10 g L−1 H2SO4, pH 1.0) and was still in a very acidic range of pH 1.0 to 1.2. The only exception was sample Y17, were the pH was with 5.0 to 5.1 much higher, most probably due to carbonate dissolution (calcite and dolomite) occurring together with the pyrite in a polymineral sample (Table 1). Based on the Fe concentrations in solution, the amount of dissolved pyrite is generally low with a highest dissolution of 2.3 % of the weighted start material (sample 13; Table 1).
The leaching mobilized about 0.84 to 88 % of REE from the samples, what is 0.33 to 11.6 μg g−1, respectively (Table 4). The 88 % were calculated for sample 8, what is rather an exception among the samples from which generally below ~30 % of REE were mobilized during leaching (Table 4). The PAAS-normalized patterns of most of the leaching samples show a common feature which is more or less pronounced (Fig. 3): a clear MREE enrichment with [Tb/La]PAAS ratios in the range of 1.1 ± 0.1 to 69.5 ± 1.1 and [Lu/Tb]PAAS ratios of 0.041 ± 0.003 to 0.89 ± 0.02. A striking MREE enrichment for example occurs in Y1a and b, 3, 4, 5, 7, 8, 12, 13, 14 and 15. Y9 and 10 are more flat or differ a bit due to additional LREE or HREE enrichment. The [MREE]PAAS value (Table 4), which was calculated to be implemented into PCA is equal or higher than 1.5 for all considered samples and with 1.5 lowest in Y9 and with 33.5 highest in Y1a. [Lu/La]PAAS ratios are except for Y9 and Y10 always higher than 1.4 indicating a HREE enrichment compared to LREE during release from pyrite. Eu anomalies occur after leaching for samples, which already had a Eu anomaly in the normalized total contents (Fig. 3).
The highest value for the R factor (Table 1), 2.49 ± 0.01, was found for Y12 indicating that more S was released or Fe was retained. An R value of 2 (1.98 ± 0.02) indicating stoichiometric dissolution was calculated for Y15. In Y14, R was 1.75 ± 0.05. For all the other samples R was always ≤1.4. In detail, for samples Y1a and b, Y8, Y9 and Y13 R values between 1.0 and 1.4 were calculated, for Y3, Y4 and Y5 much lower values of 0.1 to 0.3. In all cases these values hint on a non-stoichiometric dissolution of pyrite.
Effect of element impurities in pyrite on MREE release
The PCA carried out with released element contents (Table 3) as described above resulted in five factors with eigenvalues higher than 1 explaining in total 94.2 % of variance. The included elements split after Varimax rotation into the following factors: F1—all REE, Al; F2—Co, Ni, [MREE]PAAS; F3—Mg, Mn; F4—Pb, Zn and F5—As, Ca and Fe. Detailed values are given in Table 5. According to Einax et al. (1997), only coefficients with values of 0.7 or higher are considered.
The REE all belong together with Al to F1 due to their high geochemical similarity. Further, it becomes obvious that the intensity of MREE release ([MREE]PAAS) seems connected to the simultaneous release of Co and Ni, since all these items can be found in factor F2.
Discussion
The water samples taken in the mining-impacted areas are featured by MREE enrichment and in case of the German site a positive Ce anomaly, whereby the latter one is expected to be the result of groundwater interaction with Ce-enriched Mn-(hydr)oxides (Grawunder et al. 2010). The MREE enrichment in the water samples, regardless of whether it is groundwater (GTF), creek/river water (RZ) or lake water (SP), is a very abundant phenomenon in AMD-impacted but also naturally acidic areas as shown in this study and in literature (e.g. Gimeno Serrano et al. 2000; Johannesson and Lyons 1995; Merten et al. 2005). The source of this feature recently was still under debate. The results of the present study clearly indicate a pronounced MREE release from pyrite under acidic conditions. It is assumed that the release is process controlled, since the total content patterns are not identical to the release patterns in an acidic environment and also calculations have shown that pyrite is only partly dissolved under the applied conditions. For pyrites studied in this work the total REE contents were in a wide range (1.5 to 66.3 μg g−1) and also the amount of REE released from the pure pyrites (Tables 1 and 4) is in a large range from below 1 to almost 90 %, what is a clear indication for the fact that the incorporation of REE into pyrite might differ from sample to sample. Unfortunately, there is only little knowledge on REE incorporation into pyrite. Up to now, it was assumed to be a result of fluid inclusion reflecting the fluid composition the pyrite formed from (Mao et al. 2009) or of crystal defects (Zhao and Jiang 2007). Both hypotheses are in agreement with the total digestion results, where the total content REE patterns differ from one sample to the next due to the formation conditions and formation environment of the pyrite as far as known. REE also can be contained in nanoparticles included in the pyrite crystals as found for other trace metals (Deditius et al. 2011). The pyrite in the recent study originated from different geological environments. For instance, pyrite samples 1a and 1b were sampled in the Ronneburg area. The abundance of pyrite and marcasite in the local Palaeozoic rocks was described to vary between 0.5 to 7 % (Lange et al. 1991). Samples 1a and b, as well as 5, were all found in black shale and slate settled under marine conditions and enriched in MREE already regarding the total content. This MREE enrichment in pyrites formed under marine conditions can be explained with the model for recent MREE-enriched pore waters by Haley et al. (2004) to some degree. They stated that with increasing depth, oxygen consumption results in reducing conditions and Fe-(hydr)oxides in the sediments, which are enriched in MREE, become unstable (Haley et al. 2004). Consequently, the bound REE are released to the pore water. The formation of sulphides, including pyrite, could take over the REE pattern of the pore water. A similar statement was made by Zhao and Jiang (2007) for sulphides formed hydrothermally. In this context, another clear indication for different formation conditions is the positive Eu anomaly occurring only in some samples (e.g. TY5, TY7, TY8, TY12 and TY14). Eu anomalies, generally, can be explained by the predominant occurrence of Eu2+ under reducing conditions, or at temperatures above 250 °C and elevated pressure and thus, its higher abundance e.g. in feldspars (e.g. Nagasawa 1971; Sverjensky 1984) or even pyrite. To sum up, depending on the formation environment, the total REE patterns of pyrite can vary. However, this all might explain the different total content REE patterns, but not the similarities during the release under acidic conditions.
The pronounced MREE release from the samples is expected to be a result of the pyrite oxidation process. However, a facilitated release of MREE due to their ionic radius is unlikely; even if it was likely to substitute for Fe or S, either LREE or HREE would be released preferentially due to their especially large or small size. Redox-controlled processes also can be neglected, since only Ce and Eu would be affected to a higher extent. During pyrite oxidation, sulphate forms, which already was precluded by Johannesson and Zhou (1999) to cause MREE enrichment by complexation. Actually, sulphate is the final S-species in the pyrite oxidation process. In between, S occurs in form of various other S-species. At this point, the results of this study together with results from literature hint on a mobilization by solution complexation with another, intermediate metastable S-species, such as sulphite (SO3 2−) or thiosulphate (S2O3 2−). In fact, thiosulphate was found to be the first oxidation product of S2 2− for pyrite, but e.g. also for sphalerite (Luther 1987; Moses et al. 1987). Moses et al. (1987) also discussed that at pH < 5 thiosulphate decomposes to other S-species what later was studied by Xu and Schoonen (1995). They also attributed a key role in sulphur cycle to thiosulphate decomposing in the presence of pyrite via different pathways to sulphite and tetrathionate which depends amongst others on the pyrite surface concentration (Xu and Schoonen 1995). It is expected that even if these S-species are rather short-lived they might be important in the actual process of REE mobilization during pyrite dissolution. Unfortunately, to best knowledge, there is little information on REE complexation to these ligands, e.g. in form of speciation constants.
Previously, it was found that generally pyrite oxidation below pH 3 is a non-stoichiometric dissolution with a deficit in aqueous sulphur that is expected to precipitate as S0 or to degas as SO2 under acidic conditions (Descostes et al. 2004). However, the approach of Descostes et al. (2004) had a different experimental setting, using a higher liquid to solid ratio and HNO3 and HClO4 as elution agents with pH values around pH 2, what finally resulted in a R = [S]sol/[Fe]sol of about 1.6. They assigned this lower ratio to the disproportion of S2O3 2−, being the first S-species in pyrite oxidation, to the formation of S0 and S4O6 2−. Descostes et al. (2004) also observed R > 2 as calculated in this work for Y12 discussing that R is time dependent with a first interval of R > 2 which can last between just one up to several hours. If now neglecting the different liquid to solid ratios and the used acids, the elution solution used in the present work is more acidic (pH 1) leading to R values below 1.4. Descostes et al. (2004) discussed that at R values below 1.6 sulphite (SO3 2−) forms, which is stable under acidic conditions as SO2(g) with the option of partial degassing. Based on this information, the preferential MREE mobilization might be a result of either complexation to S2O3 2−, the first S-species in pyrite oxidation, or one of its decomposition products such as SO3 2−.
However, besides AMD-impacted or naturally acidic areas, MREE enrichment is described to occur in other environments such as pore waters in seafloor sediments (Haley et al. 2004). Indeed, the model of Haley et al. (2004) supports the hypothesis of complexation to a S-species different from SO4 2−, since it describes MREE-enriched pore water solutions in the anoxic zone, where sulphate-reducing bacteria are present (e.g. Jørgensen and Bak 1991) which also can produce ligands among the S-species causing MREE enrichment. In that case not the dissolution of MREE-enriched Fe-(hydr)oxides (Haley et al. 2004) but complexation to a S-species could cause MREE enrichment.
Finally, impurities in the pyrite samples should be taken into account. Cruz et al. (2001) stated that also the occurrence of impurities such as other sulphide minerals affects the pyrite reactivity. The pyrite samples in this study, neglecting the polymineral samples (samples 7, 10 and 17), have thoroughly non-negligible contents of Al, As, Ca and Mg, but also Co, Ni and Pb, whereby Co, Cu, Ni and Zn are in a comparable range as data presented by Descostes et al. (2004). To get an idea on the influence of the release of impurities such as Al, As, Ca, Co, Mg, Mn, Ni, Pb and Zn on the strength of MREE enrichment, a PCA was carried out. In general, the resulting factors comprise elements with similar geochemistry or geochemical distribution. For example, F1 includes the trivalent elements (REE and Al), or F4 Pb and Zn that often occur together as ores such as e.g. in the Montevecchio-Ingurtosu mining district in Italy (Medas et al. 2012). However, the more interesting part was the relation of Ni, Co and MREE enrichment, all included in factor F2. As previously found by Lehner and Savage (2008) working with Co and Ni doped synthetic pyrites, these two elements increase the oxidation rate of pyrite slightly under acidic conditions (pH 2). Thus, factor F2 seems to comprise items and elements having a connection with the oxidation rate of pyrite. Since also [MREE]PAAS belongs to this factor this can be seen as a hint that the strength of MREE enrichment is connected to the oxidation rate as well. As discussed above, a metastable S-species is expected to cause MREE enrichment in general. A higher oxidation rate due to impurities such as Ni or Co (Lehner and Savage 2008) might also increase the release of MREE by higher concentrations of this metastable S-species.
Conclusions
Based on the recent data the occurrence of MREE enrichment in AMD-impacted areas is assumed to be most probably connected to complexation to an intermediate S-species during pyrite oxidation. Calculation of molar [S]sol/[Fe]sol ratios indicate non-stoichiometric dissolution and the presence of SO3 2−, but based on the current data also other S-species such as S2O3 2− influencing the REE release cannot be excluded. The strength of the MREE enrichment most probably can be traced back to different oxidation rates of pyrite, due to its impurities. Complexation constants for REE and intermediate S-species in pyrite oxidation were not determined up to now, but this complexation with such a species might probably be the missing link to explain MREE enrichment in AMD- impacted but also naturally acidic areas.
References
Assarsson G, Grundulis V (1961) Chemical investigations of upper Cambrian shales at Hynneberg, Närke. Geol Fören Förhandl 83:269–277
Bau M, Dulski P (1996) Distribution of yttrium and rare-earth elements in the Penge and Kuruman iron-formations, Transvaal Supergroup South Africa. Precambrian Res 79:37–55
Bau M, Möller P (1992) Rare earth element fractionation in metamorphogenic hydrothermal calcite, magnesite and siderite. Mineral Petrol 45:231–246
Bozau E, Leblanc M, Seidel JL, Stärk HJ (2004) Light rare earth elements enrichment in an acidic mine lake (Lusatia, Germany). Appl Geochem 19:261–271
Cook NJ, Ciobanu CL (2004) Bismuth tellurides and sulphosalts from the Larga hydrothermal system, Metaliferi Mts, Romania: paragenesis and genetic significance. Mineral Mag 68:301–321
Cook NJ, Ciobanu CL, Pring A, Skinner W, Shimizu M, Danyushevsky L, Saini-Eidukat B (2009) Trace and minor elements in sphalerite: a LA-ICP-MS study. Geochim Cosmochim Acta 73:4761–4791
Cruz R, Bertrand V, Monroy M, González I (2001) Effect of sulfide impurities on the reactivity of pyrite and pyritic concentrates: a multi-tool approach. Appl Geochem 16:803–819
Deditius AP, Utsunomiya S, Reich M, Kesler SE, Ewing RC, Hough R, Walshe J (2011) Trace metal nanoparticles in pyrite. Ore Geol Rev 42:32–46
Descostes M, Vitorge P, Beaucaire C (2004) Pyrite dissolution in acidic media. Geochim Cosmochim Acta 68:4559–4569
Duma S (2009) The impact of mining activity upon the aquatic environment in the southern Apuseni Mountains. Rom Rev Reg Stud 5:51–66
Einax JW, Zwanziger HW, Geiß S (1997) Chemometrics in environmental analysis. Wiley-VCH, Weinheim
Elderfield H, Upstill-Goddard R, Sholkovitz ER (1990) The rare earth elements in rivers, estuaries, and coastal seas and their significance to the composition of the ocean waters. Geochim Cosmochim Acta 54:971–991
García MG, Lecomte KL, Pasquini AI, Formica SM, Depetris PJ (2007) Sources of dissolved REE in mountainous streams draining granitic rocks, Sierras Pameanas (Córdoba, Argentina). Geochim Cosmochim Acta 71:5355–5368
Gimeno Serrano MJ, Sanz LFA, Nordstrom DK (2000) REE speciation in low-temperature acidic waters and the competitive effects of aluminum. Chem Geol 165:167–180
Grandjean-Lécuyer P, Feist R, Albarede F (1993) Rare-earth elements in old biogenic apatites. Geochim Cosmochim Acta 57:2507–2514
Grawunder A, Lonschinski M, Merten D, Büchel G (2009) Distribution and bonding of residual contamination in glacial sediments at the former uranium mining leaching heap of Gessen/Thuringia, Germany. Chem Erde 69(S2):5–19
Grawunder A (2010) Hydrogeochemistry of rare earth elements in an acid mine drainage influenced area. Dissertation, Friedrich Schiller University, Jena
Grawunder A, Lonschinski M, Boisselet T, Merten D, Büchel G (2010) Hydrogeochemistry of rare earth elements in an AMD-influenced area. – In: Wolkersdorfer Ch, Freund A (Eds): Mine Water & Innovative Thinking. Sydney, Nova Scotia (CBU Press), p. 347 – 351
Grawunder A, Merten D (2012) Rare earth elements in acidic systems—biotic and abiotic impacts. In: Kothe E, Varma A (Eds) Bio-geo interactions in metal-contaminated soils. Soil Biology Vol. 31, Springer, Berlin, p. 81–97
Haley BA, Klinkhammer GP, McManus J (2004) Rare earth elements in pore waters of marine sediments. Geochim Cosmochim Acta 68:1265–1279
Hannigan RE, Sholkovitz ER (2001) The development of middle rare earth element enrichments in freshwaters: weathering of phosphate minerals. Chem Geol 175:495–508
Ichikuni M (1960) Sur la dissolution des minerais sulfurés en divers milieu. II. Dissolution de la pyrite et de la chalcopyrite. Bull Chem Soc Jpn 33:1052–1057 (in French)
Johannesson KH, Lyons WB, Yelken MA, Gaudette HE, Stetzenbach KJ (1996) Geochemistry of the rare-earth elements in hypersaline and dilute acidic natural terrestrial waters: complexation behavior and middle rare-earth element enrichments. Chem Geol 133:125–144
Johannesson KH, Lyons WB (1995) Rare-earth element geochemistry of Color Lake, an acidic freshwater lake on Axel-Heiberg-Island, Northwest-Territories, Canada. Chem Geol 119:209–223
Johannesson KH, Zhou XP (1997) Geochemistry of the rare earth elements in natural terrestrial waters: a review of what is currently known. Chin J Geochem 16:20–42
Johannesson KH, Zhou XP (1999) Origin of middle rare earth element enrichments in acid waters of a Canadian High Arctic lake. Geochim Cosmochim Acta 63:153–165
Jones A, Rogerson M, Greenway G, Potter HAB, Mayes WM (2013) Mine water geochemistry and metal flux in a major historic Pb-Zn-F orefield, the Yorkshire Pennines, UK. Environ Sci Pollut Res 20:7570–7581
Jørgensen BB, Bak F (1991) Pathways and microbiology of thiosulfate transformations and sulfate reduction in a marine sediment (Kattegat, Denmark). Appl Environm Microbiol 57:847–856
Karlsson S, Sjöberg V, Pourjabbar, A, Grandin A and Allard B (2012) Distribution of rare earth elements and other metals in a stratified acidic pit lake in black shales 45 years after mine closure. In: Price WA, Hogan C, Tremblay G (Eds) Proceedings 9th International Conference on Acid Rock Drainage (ICARD) 2012; Ottawa, Canada (CD-ROM, 12p)
Lange G, Mühlstedt P, Freyhoff G, Schröder B (1991) Der Uranerzbergbau in Thüringen und Sachsen—ein geologisch bergmännischer Überblick. Erzmetall 44:162–171 (in German)
Lécuyer C, Reynard B, Grandjean P (2004) Rare earth element evolution of Phanerozoic seawater recorded in biogenic apatites. Chem Geol 204:63–102
Lehner S, Savage K (2008) The effect of As, Co, and Ni impurities on pyrite oxidation kinetics: batch and flow-through reactor experiments with synthetic pyrite. Geochim Cosmochim Acta 72:1788–1800
Luther GW (1987) Pyrite oxidation and reduction: molecular orbital theory considerations. Geochim Cosmochim Acta 51:3196–3199
Mao G, Hua R, Gao J, Li W, Zhao K, Long G, Lu H (2009) Existing forms of REE in gold-bearing pyrite of the Jinshan gold deposit, Jiangxi Province, China. J Rare Earths 27:1079–1087
McLennan SM (1989) Rare earth elements in sedimentary rocks: influence of provenance and sedimentary processes. In: Lipin BR, McKay GA (Eds) Geochemistry and mineralogy of rare earth elements. Rev Min Geochem 21:169–200
McLennan SM (1994) Rare earth element geochemistry and the “tetrad” effect. Geochim Cosmochim Acta 58:2025–2033
Medas D, Cidu R, Lattanzi P, Podda F, De Giudici G (2012) Natural biomineralization in the contaminated sediment–water system at the Ingurtosu abandoned mine. In: Kothe E, Varma A (eds) Bio-Geo interactions in metal-contaminated soils. Soil Biology Vol. 31, Springer, Berlin, pp 113–130
Merten D, Geletneky J, Bergmann H, Haferburg G, Kothe E, Büchel G (2005) Rare earth element patterns: a tool for understanding processes in remediation of acid mine drainage. Chem Erde 65(S1):97–114
Morgan JW, Wandless GA (1980) Rare earth element distribution in some hydrothermal minerals: evidence for crystallographic control. Geochim Cosmochim Acta 44:973–980
Moses CO, Nordstrom DK, Herman JS, Mills AL (1987) Aqueous pyrite oxidation by dissolved oxygen and by ferric iron. Geochim Cosmochim Acta 51:1561–1571
Nagasawa H (1971) Partitioning of Eu and Sr between coexisting plagioclase and K-feldspar. Earth Planet Sci Lett 13:139–144
Neubauer F, Lips A, Kouzmanov K, Lexa J, Ivǎşcanu P (2005) 1: Subduction, slab detachment and mineralization: the Neogene in the Apuseni Mountains and Carpathians. Ore Geol Rev 25:13–44
Pérez-López R, Gelgado J, Nieto JM, Márquez-García B (2010) Rare earth element geochemistry of sulphide weathering in the São Domingos mine area (Iberian Pyrite Belt): a proxy for fluid–rock interaction and ancient mining pollution. Chem Geol 276:29–40
Reynard B, Lécuyer C, Grandjean P (1999) Crystal-chemical controls on rare-earth element concentrations in fossil biogenic apatites and implications for paleoenvironmental reconstructions. Chem Geol 155:233–241
Smedley PL (1991) The geochemistry of rare-earth elements in groundwater from the Carnmenellis Area, Southwest England. Geochim Cosmochim Acta 55:2767–2779
Sverjensky DA (1984) Europium redox equilibria in aqueous solution. Earth Planet Sci Lett 67:70–78
Verplanck PL, Nordstrom DK, Taylor HE, Kimball BA (2004) Rare earth element partitioning between hydrous ferric oxides and acid mine water during iron oxidation. App Geochem 19:1339–1354
Welch SA, Christy AG, Isaacson L, Kirste D (2009) Mineralogical control of rare earth elements in acidic sulfate soils. Geochim Cosmochim Acta 73:44–64
Wismut GmbH (1994) Sanierungskonzept Standort Ronneburg. Stand Dezember 1994. Internal Report, Wismut GmbH, Chemnitz (in German)
Xu Y, Schoonen MAA (1995) The stability of thiosulfate in the presence of pyrite in low-temperature aqueous solutions. Geochim Cosmochim Acta 59:4605–4622
Zhao KD, Jiang SY (2007) Rare earth element and yttrium analyses of sulfides from the Dachang Sn-polymetallic ore field, Guangxi Province, China: implication for the ore genesis. Geochem J 41:121–134
Acknowledgements
The authors thank the project “Umbrella” funded by the EU (FP7-ENV-2008-1 no. 226870) under the coordination of E. Kothe (Friedrich Schiller University Jena) for financial support. Further, we thank I. Kamp, G. Rudolph and G. Weinzierl (all Friedrich Schiller University Jena) for ICP-OES, DOC and IC measurements, as well as all partners within the Umbrella project, especially the Swedish group (B. Allard, M. Bäckström, S. Karlsson, V. Sjöberg; University of Örebro) and the Romanian group (A. Neagoe, V. Iordache; University of Bucharest) for enabling access to the field sites and supporting us during field work, and G. De Giudici (University of Cagliari) for helpful comments. We also thank an anonymous reviewer for helpful comments improving the manuscript once more. Furthermore, we are grateful to A. Kötschau (Friedrich Schiller University Jena) for support on statistical analysis, as well as B. Kreher-Hartmann from the Mineralogical Collection of the Friedrich Schiller University Jena, S. Meißner and M. Riefenstahl for providing further mineral samples and F. Schäffner (Friedrich Schiller University Jena) as well as P. T. Stancu (University of Bucharest) for their help during field work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article
Cite this article
Grawunder, A., Merten, D. & Büchel, G. Origin of middle rare earth element enrichment in acid mine drainage-impacted areas. Environ Sci Pollut Res 21, 6812–6823 (2014). https://doi.org/10.1007/s11356-013-2107-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-013-2107-x