
Abstract
The aim of the present investigation was to exploit the high specific surface area of activated carbons in immobilizing the manganese and iron oxides as to obtain a suitable, efficient and cost effective and environment benign wastewater treatment process in the remediation of cadmium-contaminated waters. The manganese and iron oxides were impregnated in situ onto the surface and pores of the activated carbons precursors to the rice hulls and areca nut wastes. The solids were characterized with the help of Fourier transform infrared spectroscopy and X-ray diffraction analytical data, and the BET specific surface area as obtained. The surface morphology of these solids was discussed with the help of scanning electron microscopic images. The activated carbon samples along with the manganese and iron immobilized activated carbons were further employed in the batch and column reactor operations in the remediation of cadmium-contaminated waters. The batch data showed that an increase in sorptive pH from 2.0 to 10.0 and concentration from 1.0 to 20 mg/L favoured the uptake of cadmium by these solids. Moreover, the 1,000 times increase in background electrolyte concentrations NaNO3 caused an insignificant decrease in cadmium uptake by these solids, which inferred that sorbing ions/species were sorbed specifically and forming ‘inner-sphere’ complexes onto the solid surface. The concentration dependence data were utilized to model various adsorption isotherms and indicated that Freundlich adsorption isotherm was reasonably fitted well. The kinetic data was fitted well to the pseudo-second-order rate equations; hence, the equilibrium sorption capacity was estimated. Furthermore, the dynamic experiments carried out by the column experiments and the breakthrough data were fitted well to the non-linear Thomas equations; accordingly, the loading capacity of the column was estimated. Iron or manganese immobilized activated carbons showed relatively higher loading capacity compared to its precursor activated carbons hence showing its possible implication in the remediation processes. Moreover, among these modified ACs, IIAC showed higher removal capacity than the MIAC solid.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cadmium is one of the transition element which showed severe occupational and environmental health hazards towards the human being (Jarup and Akesson 2009; Waalkes 2003). The biological half-life of cadmium was reported to be extremely high, which makes a cumulative toxic effect within the bio system (Goering et al. 1994). Cadmium primarily binds to metallothionein (MT), a metal-binding protein within the liver and kidney (Goering et al. 1994; Klassen et al. 1999). Although cadmium competes with zinc, upon binding to MT, however, excess zinc dose may antagonize several bio-toxic effects of cadmium including tumour formation (Waalkes 2000). Long-term exposure to cadmium exhibited osteomalacia and osteoporosis along with kidney damage (Jarup and Akesson 2009). Moreover, studies indicated that even in the absence of excessive cadmium-induced renal tubular dysfunction, low-level environmental exposure seemingly causes calciuria, having reactive changes in calciotropic hormones (Schutte et al. 2008). The study related to its carcinogenicity; it was recognized as human carcinogen (group 1), referred particularly for the occupationally exposed persons (IARC International Agency for Research on Cancer 1993). Moreover, cadmium intake may cause prostatic proliferative lesions, including adenocarcinomas as studied on rats (Waalkes 2000; Waalkes et al. 1999). It was reported that excessive inhalation of cadmium causes pulmonary adenocarcinomas, etc. (Waalkes and Misra 1996).
Cadmium enters into the environment by the deposition of airborne cadmium, processing of cadmium-containing ores, use of numerous cadmium-containing fertilizers and sewage sludge contaminating, predominantly, the soils, and it is recycled through the vegetables/plants then to human or animal. In absence of smoking, the environmental cadmium is the main source of cadmium uptake by the un-occupational human. Interestingly, more than 80 % of the cadmium in food comes from cereals, vegetables and potato (Olsson et al. 2002), and on average, one receives ca. 8 to 25 μg cadmium per day through food stuff (Egan et al. 2007; Larsen et al. 2002; Olsson et al. 2002). The other common industrial sources of cadmium were reported to be electroplating, smelting, alloy manufacturing, pigments, plastic industry, cadmium-containing batteries, mining and refining industries, nuclear reactor shields, rods etc.(Amoyaw et al. 2009; Benguell and Benaissa 2002). Because of its acute toxicity, the maximum permissible limit in drinking water was reduced to the level of 0.003 ppm as directed by the Government of India (GOI 2008) or World Health Organization (WHO 2008). The low limit of cadmium as prescribed by the regulatory agencies indicated accordingly the low level removal of cadmium from aquatic environment. In line with this, the commonly employed techniques, viz. precipitation, co-precipitation, nanofiltration, ultrafiltration, reverse osmosis, solvent extraction, oxidation, ion-exchange, adsorption etc. (Berbenni et al. 2000; Mulligan et al. 2001; Pandey et al. 2008), were possible methods of treatment. These methods were not only useful in the remediation of heavy metal toxic ions but also showed enhanced applications in the removal of variety of water pollutants including dyes and other potential contaminants (Gupta et al. 2011a; Grassi et al. 2012; Liu et al. 2011; Mittal et al. 2008; Xu et al. 2009). The precipitation or co-precipitation methods were found to be inappropriate in the trace level removal of heavy metals. Moreover, the coexistence of alkali-earth metals lies in much higher concentrations and made additional environmental load (Vilensky et al. 2002; Zhu et al. 2007). Nevertheless, the adsorption with ion-exchange process using a variety of materials including inorganic, biomasses or even waste materials was reported to be possible alternative methods, but the suitability and efficiency of the materials were one of the major issues in such treatment processes at the low level removal of contaminants (Gupta et al. 2009, 2011b; Mishra et al. 1998a).
The efficiency of 20 different species of plant leaves was optimized in the cadmium removal and showed that styrax, plum, pomegranate and walnut leaves possessed relatively higher removal capacity (Salim et al. 2008). Similarly, the olive leaves were utilized in the removal of Cd(II). The process of sorption was conducted under sonication with continuous stirring as to achieve an enhanced Cd(II) removal capacity (Hamdaoui 2009). The chitin (Benguell and Benaissa 2002) and chitosan-based composite material precursor to activated carbon (Hydari et al. 2012) showed very high removal capacity for cadmium. The removal capacity of cadmium was found to be 52.63 mg/g using composite materials (Hydari et al. 2012). Similarly, the fast and parts per billion level removal of Cd(II) from aqueous solutions was proposed by employing the poly[4-(4-vinylbenzyloxy)-2-hydrobenzaldehyde], a salicylaldehyde chelating functional group-based polymer (Amoyaw et al. 2009). The nanofibre composite material poly(vinyl alcohol)/tetraethylorthosilicate/aminopropyltriethoxysilane was obtained by the sol–gel/electro-spinning process and was further assessed for the removal of cadmium (Irani et al. 2012). There were a variety of biosorbents suggested for the effective removal of Cd(II) from aqueous solution under the active or passive uptake process including living cells of microalgae Tetraselmis suecica (Perez-Rama et al. 2002); pre-treated fungal biomass of Rhizopus oryzae, Rhizopus oligosporus, Aspergillus orzae and Rhizopus arrhizus (Yin et al. 1999); Oscillatoria sp. H1 (Cyanobacteria, microalgae, immobilized live and heat-inactivated; Katircioglu et al. 2008); peels of peas, broad bean, medlar and fig leaves (Benaissa 2006); Calotropis procera (a perennial wild plant) etc. (Pandey et al. 2008). Similarly, the electroplating waste material was found to be potential sorbent material in the Cd(II) removal from the aqueous solutions, and the metal-laden waste was suggested to be immobilized with cement for ultimate disposal (Bhatnagar and Minocha 2009). Thermodynamics of adsorption of Cd(II) onto the China clay from aqueous solutions was investigated, and it was suggested that the isosteric heat of adsorption, ΔH X, was decreased with decreasing the value of surface coverage (Sharma 2008). The microporous titanosilicate ET-4 (Engelhard titanium silicates No. 4; Barreira et al. 2009) or nano-zero-valent iron particles (Boparai et al. 2011) found a kind of new materials in the treatment process. The manganese-loaded resin (Zhu et al. 2007), manganese dioxide impregnated sand (Lee et al. 2010) or even iron oxide nanoparticles immobilized sand (Lee et al. 2012) materials have shown their efficiency in the remediation of water contaminated with Cd(II). These studies showed the varied applicability of the materials in the remediation of Cd(II)-contaminated waters; the present work is an extension of previous studies immobilizing the manganese dioxide or iron oxide nanoparticles onto the sand substrate which showed effective treatment materials for Cd(II) decontamination. The objective of the present study is to exploit the use of relatively high specific surface area of activated carbon precursors to the rice hulls and areca nut waste materials by immobilizing the manganese and iron oxide particles onto the surface and pores of it, and were further employed in the removal of Cd(II) from aquatic environment under the batch and column reactor operations. The study was intended to provide, perhaps, a cost-effective and cleaner treatment process in the remediation of aquatic environment contaminated with cadmium.
Materials and methods
Materials
The dead biomasses viz. rice hulls and the areca nut wastes were collected from Aizawl, Mizoram, India. Sulphuric acid and ammonia were obtained from Merck, India. The manganese nitrate as Mn(NO3)2.6H2O, 97 % extra pure, was obtained from Junsei Chemical Co. Ltd., Japan, and cadmium(II) sulphate and iron(III) nitrate enneahydrate were obtained from Kanto Chemicals Co. Inc., Japan. Sodium hydroxide, GR Grade, and nitric acid were obtained from Duksan Pure Chemicals Co. Ltd., Korea. The de-ionized water was further purified by the Millipore water purification system (Milli-Q+).
Preparation of activated carbons (AC)
The raw biomass materials, i.e. rice husk and areca nut wastes, were washed with de-ionized distilled water to remove any adhering impurities. Samples were dried at room temperature, and these solids were digested in concentrated H2SO4, and the content was kept at 120 °C for 2 h. The carbonized carbon obtained was washed with distilled water until the filtrate solution was reached to pH ∼ 4.0. Again, it was dried at 70 °C, and the samples were titrated with small volume of NH3 solution to neutralize excess acids. The sample was washed with distilled water and completely dried at 70 °C. It was cooled at room temperature and grounded to obtain fine powders. Furthermore, this carbonized carbon was activated by using muffle furnace at 800 °C in N2 environment for 6 h, and the activated carbon obtained from rice husk [AC(R)] and areca nut waste [AC(N)] were used for batch experiment, as well as for the preparation of manganese or iron oxide immobilized activated carbons [MIAC(R), MIAC(N), IIAC(R) or IIAC(N)].
Preparation of manganese or iron oxide immobilized activated carbon
The manganese oxide or iron oxide immobilized activated carbon (MIAC or IIAC) was obtained by taking 60 g of AC in a round bottom flask with 100 mL of 0.025 M manganese nitrate solution or iron nitrate solution (pH ∼ 9) and was kept in a rotary evaporator at 60 °C at the rotating speed of 30 rpm. In addition, almost 90 % of the water was removed by applying slowly the vacuum. The slurry was taken out in a beaker and kept in a drying oven at 90 °C to dry the solids completely. Furthermore, the sample was kept for 2 h at 110 °C for the stabilization of immobilized manganese oxide or iron oxide. Samples were taken out from the drying oven, cooled at room temperature and washed with distilled water. It was dried again at 70 °C. These samples were then used for further investigations.
Characterization of the materials
The solids obtained were characterized by Fourier transform infrared spectroscopy (FT-IR; Tensor 27, Bruker, Billerica, MA, USA, by KBR disk method) and X-ray diffraction (XRD machine, i.e. PANalytical, Netherland; Model X'Pert PRO MPD). The XRD data was collected having the scan rate of 0.034 of 2θ illumination at an applied voltage of 45 kV with the observed current of 35 mA, using CuK α radiations having a wavelength of 1.5418 Å. The BET specific surface area was obtained by the Protech Korea BET surface area Analyzer (Model ASAP 2020). The surface morphology of these solids was obtained by the scanning electron microscope machine (SEM model: FE-SEM-Model, SU-70, Hitachi, Japan). Furthermore, the amount of manganese or iron contents contained by ACs, MIACs and IIACs was determined by the standard US EPA (3050B) method.
Stability of coatings
The solids MIAC or IIAC were subjected to stability tests conducted at a wide range of pH values (pH 2.0 to 10.0). For the purpose of this study, 100 mL of water samples was taken in polyethylene bottles, and the pH of these water samples was adjusted from pH 2.0 to 10.0 by the addition of drops of concentrated HNO3 or NaOH solutions. Of solids, 0.25 g, i.e. ACs, MIACs or IIACs, was then introduced. The samples were kept at 25 °C in an automatic shaker for 24 h. The solutions were filtered with 0.45-μm syringe filter and subjected to total manganese or total iron concentrations using atomic absorption spectrometer (Fast Sequential Atomic Absorption Spectrometer, Model AA240FS, Varian).
pHpzc determination of the solids
The pHpzc (point of zero charge) was determined using the known method as described earlier (Hydari et al. 2012; Irani et al. 2012). Of double distilled water, 500 mL was added to an Erlenmeyer flask, capped with cotton. It was slowly but continuously heated until boiling for 20 min as to expel the dissolved CO2. The flask was capped immediately to prevent re-absorption of atmospheric CO2 by water. Then, 50 mL of 0.01-M NaCl solutions was prepared from CO2 free water and taken in two different bottles. The pH of each solution in each flask was adjusted to pH values of 2, 4, 6, 8, 10 and 12 by adding 0.1 M HCl or 0.1 M NaOH solutions. Of the solid sample, 0.15 g was then introduced in each bottles, and the final pH was measured after 24 h which was agitated at 25 °C. The pHpzc was taken as the point at which the curve crossed the line; pHfinal equals to pHinitial.
Batch reactor operations
Batch experiments were carried out to obtain the adsorption data with the variation of sorptive pH, contact time, initial metal concentration and background electrolyte concentrations. The adsorption of Cd(II) was investigated by taking 0.25 g of ACs/MIACs/IIACs solids separately in 100 mL of sorptive solution. The solution mixture was equilibrated by using automatic shaker (KUKJE, Shaking Incubator, Korea model 36-SIN-125) for 24 h at 25 ± 1 °C temperature. The solution was then filtered by 0.45-μm syringe filter; the final pH was measured and reported. The filtered solutions were subjected for AAS analysis to obtain the final metal concentrations. Adjustment of the pH was done by the drop-wise addition of concentrated HNO3 or concentrated NaOH. The time dependence data was collected at different time intervals having a Cd(II) concentration of 10.0 mg/L and at constant pH 5.0 and temperature 25 ± 1 °C. Similarly, the concentration dependence data was obtained by varying the initial sorptive concentration from 1.0 to 20.0 mg/L, and other parameters were kept constant, viz. pH 5.0 and temperature 25 ± 1 °C.
Column studies
The column experiments were performed using a glass column (1 cm inner diameter) packed with 1.0 g of ACs/MIACs/IIACs (kept middle in the column); below and above to this, 1.0 g each of bare sand (30–60 BSS in size) was kept, and then it was packed with glass beads. The sorptive solution was pumped upward from the bottom of the column using Acuflow Series II high-pressure liquid chromatograph at a constant flow rate of 1.00 mL/min. Effluent samples were then collected using Spectra/Chrom CF-2 fraction collector. The collected samples were again filtered with 0.45-μm syringe filter, and the total bulk sorptive concentration was measured using AAS.
Results and discussion
Characterization of materials
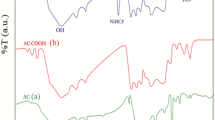
FT-IR data were obtained for the activated carbons obtained by the rice hulls and areca nut, along with the manganese and iron oxide immobilized activated carbons, and the results were presented graphically in Fig. 1a, b, respectively, for the areca nut and rice hulls. Carbon samples could possess various functional groups having different surface structures, viz. aliphatic, aromatic and cyclic, since the IR bands were observed at around 1,480 cm−1 and regions around 1,110 cm−1 for both these ACs. Predominant and broad peak appeared at around 3,470–3,495 and were ascribed to the stretching vibration of –OH groups (carboxyl, alcohol or phenols) etc., or it may be attributed to the absorption of some atmospheric water during FTIR measurements (Gun'ko et al. 2005; Gupta et al. 2011b). However, it was reported that IR bands that occurred around 3,300 and 3,250 cm−1 were indicative of the hydroxyl groups, but usually, the band is shifted at around 3,500 cm−1 in case of non-bonded alcohols, phenols and carboxylic acids (Gomez-Serrano et al. 1996). Furthermore, peaks in between 2,930 and 2,910 cm−1 were ascribed to the C-H stretching and C-H scissoring bands, respectively (Gupta et al. 2011b). Moreover, it was suggested that the peaks at 2,800–2,900 cm−1 indicated the existence of aldehyde groups (Gun'ko et al. 2005). The weak peaks occurring at around 1,460 cm−1 showed the C-H vibration in –CH2- deformation (Puziy et al. 2002). Moreover, the bands that occurred in between 1,600 and 1,400 (more predominant in the areca nut ACs) could be assigned as C = C vibrations. It was pointed out that the peaks in the region 1,350 and 900 cm−1 are because of the C-O or C = O vibrations (Al Bahri et al. 2012). Similarly, a vibration band occurring around 870 cm−1 was obtained because of external bending of –C-H for different substituted benzene ring (Kong et al. 2013). The peak at around 450–475 cm−1 was assigned as the stretching vibrations of Fe-O (Gupta et al. 2011b; Rodulfo-Baechler et al. 2004). This peak was present in both IIACs samples as well with less intensity indicating that the bare ACs also included some iron oxide, which was again confirmed by the digestive analysis conducted separately. Similarly, peaks at around 840 cm−1 inferred to the υ s vibrations of Si-O-Si or δ-Fe-OH predominant in the rice hulls activated carbon samples (Li et al. 2010). On the other hand, Mn-O stretching vibrations usually occurred at 940 cm−1 (Nakamoto 1997), but none of the samples showed peak in this region suggesting that the content of manganese was too low; hence, the vibration intensity was almost negligible in this region.

FT-IR data for the AC, MIAC and IIAC solids: (a) areca nut and (b) rice hulls
Furthermore, these solids were subjected to XRD analysis, and the results were presented graphically in Fig. 2. It was observed that activated carbon of rice hulls possessed diffraction peak at 2θ value of 22.5, which showed a characteristic peak of amorphous silica (Liu et al. 2012). Similarly, the AC of areca nut showed very broad diffraction peak almost in the same region indicating perhaps the amorphous silica. The XRD data further indicated that ACs have no ordered crystalline structure since no sharp reflections were observed for these samples. A similar XRD pattern was obtained for the IIAC(R) or IIAC(N) peak at the 2θ value of 39, which was indicative of akaganeite (β-FeOOH; Vitela-Rodriguez and Rangel-Mendez 2013). Possibly the other diffraction peaks of the akaganeite at 2θ value of 35 were missing because of low content of iron or weak diffraction at this 2θ value (Depci 2012). On the other hand, the MIACs samples could not show sharp diffraction peaks in the XRD pattern except a weak diffraction peak at the 2θ value of 32.5 [MIAC(R)] possibly due to the presence of Mn3O4 mineral phase (Si et al. 2009). No other characteristic peaks were observed in the MIACs samples. This was perhaps because of the low content of impregnated manganese onto the solid ACs surfaces. Previously, it was reported that no significant XRD peak was observed with the manganese or iron impregnated solids using different substrates because the amorphous iron or manganese was deposited, or the particles were significantly small to diffract or because of very low content of iron/manganese (Corapcioglu and Huang 1987; Depci 2012; Muniz et al. 2009; Tiwari et al. 2011).

XRD data for various samples of ACs, IIACs and MIACs
The BET specific multipoint surface area was obtained and found to be 379.77, 322.14, 370.88, 303.79, 371.04 and 315.83 m2/g, respectively, for AC(N), AC(R), IIAC(N), IIAC(R), MIAC(N) and MIAC(R) solids. The significant specific surface area obtained for the AC samples was, therefore, employed for the impregnation of iron or manganese oxides onto these solids. The specific surface area of AC was slightly decreased in the presence of iron or manganese oxides since the iron or manganese occupied the spaces and hence decreased the specific surface area. However, the presence of iron or manganese oxides enabled the solids towards the effective and efficient removal of cadmium from aqueous solution. The similar decrease in specific area was reported previously in the case of iron-doped activated carbon sample (Fierro et al. 2009), whereas the manganese-impregnated sand showed slightly enhanced specific surface area compared to the bare sand because of the very compact structure of sand (Lee et al. 2010).
The SEM images of AC(N), AC(R), MIAC(N), MIAC(R), IIAC(N) and IIAC(R) were shown in Fig. 3. It was reported that the nano- or small-sized manganese oxides exhibit an adsorption superior to its bulk counterpart because of its polymorphic structures and higher specific surface area (Wang et al. 2011). The SEM images clearly indicated that the surface morphology was changed significantly with manganese- or iron-modified activated carbon samples. Surface structure of AC(R) showed very porous surface, and the pores were very unevenly distributed on the surface but not fully opened on the surface. It was observed that the silicon oxide mostly aggregated on the pores or surface of AC(R). The pore size varies significantly. Compared with AC(R), the AC(N) possessed more porous surface. Moreover, the pores on the AC(N) surface were partly opened but were, however, mostly distributed evenly on the surface. Furthermore, the SEM images of MIAC and IIAC showed that the manganese dioxide or iron oxide particles were significantly aggregated or even clustered onto the surface of AC samples or even within the pores of the ACs. The particles were distributed unevenly and predominantly small in size. Earlier reports indicated that the nano-sized manganese dioxide or iron oxide particles were very orderly aggregated onto the sand surface providing distinct specific surface area (Lee et al. 2010, 2012). These results, therefore, indicated that the manganese oxide or iron oxide particles were significantly immobilized onto the surface of AC(N) and AC(R). Furthermore, the effective amount of manganese or iron content of the samples was analyzed by US EPA 3050B method. The manganese content was found to be 66.0, 56.0, 2,055.0 and 2,331.0 mg/kg, respectively, for the solids AC(N), AC(R), MIAC(N) and MIAC(R). Similarly, the iron content was found to be 107.0, 39.0, 2,442.0 and 2,221.0 mg/kg, respectively, for the solids AC(N), AC(R), IIAC(N) and IIAC(R).

SEM images of the ACs and modified ACs
Stability of MIAC and IIAC
The stability test results were shown in Fig. 4. It is evident that the manganese or iron immobilization was fairly stable within the pH range from 3.0 to 10.0 since no significant amount of the manganese or iron was leached in the bulk solution. These results further pointed out that the solids possessed wider applicability in the treatment of water samples in the pH range from 4.0 to 10.0. The similar stability of iron or manganese coating was obtained for the sand samples and indicated the utility of the solids in the wastewater treatment technology (Lee et al. 2010, 2012).

Stability of manganese or iron on the impregnated samples of ACs as a function of solution pH
Batch reactor operations
Effect of pH
The change in solution pH affects the surface charge of the adsorbents; therefore, the effect of pH is an important parameter in helping to deduce the mechanism involved at the solid/solution interface (Momcilovic et al. 2011; Tiwari et al. 2007). The sorption data was obtained by varying the initial pH from 2.0 to 10.0 and at constant initial Cd(II) concentration of 10.0 mg/L. The results obtained were presented graphically in Fig. 5a, b, respectively, for AC(N)- and AC(R)-based solids. The figures clearly indicated that, in general, increasing the pH from 2.0 to 10.0 of the sorptive solution caused a gradual increase in the adsorption of Cd(II) by these solids. More precisely, it was noted that increasing the pH from 2.39 to 10.11 for AC(N), 2.58 to 10.23 for MIAC(N) and 2.00 to 9.69 for IIAC(N) caused an increase in Cd(II) uptake, respectively, from 8.15 % to 100.0 % for AC(N), 13.24 % to 100 % for MIAC(N) and 31.23 % to 100 % for IIAC(N). Similarly, increasing the pH from 2.29 to 10.18 for AC(R), 2.57 to 10.38 for MIAC(R) and 205 to 9.30 for IIAC(R) caused an increase in Cd(II) removal, respectively, from 10.90 % to 100.0 %, 15.38 % to 100.0 % and 36.26 % to 100.0 %. This could be explained with the help of the surface properties of solid surface along with the speciation of sorbing species. Speciation studies demonstrated elsewhere (Lee et al. 2012) indicated that the cadmium exists as a positively charged Cd2+ from low pH to pH 8.8, and beyond that, the cadmium converted into Cd(OH)2 (S) species which occurs 100 % at pH 9.4. However, in between the Cd2+ and Cd(OH)2(S), Cd(OH)+ also exists as one of the insignificant species with maximum of 4.8 % at pH 8.8. In other words, in a wide range of pH, cadmium exists in cationic form and carries net positive charge. On the other hand, the pHpzc of the adsorbents was found to be 6.7, 6.6, 6.5, 6.5, 6.2 and 6.1, respectively, for the solids AC(N), AC(R), MIAC(N), MIAC(R), IIAC(N) and IIAC(R). This suggested that below this pH, the surface of these solids carries a net positive charge, and beyond this, it contains a net negative charge. Therefore, the lower value of sorption occurred at lower pH because the electrostatic repulsion took place between the Cd2+ and the positively charged solid surface. Moreover, at high acidic conditions, i.e. at very low pH around pH 2–3, perhaps a competition could occur between the excess of hydrogen ions and metal ions towards the same sites of solid surface which further suppressed the Cd(II) adsorption. Increasing the pH caused for a decrease in H+ ion concentrations due to successive de-protonation. This resulted in gradual increase in negative charge density onto the solid surface, which enables to favour more adsorption of the metal ions (Zhu et al. 2007). The observed data supported the modelling and experimental data obtained to analyse the surface properties of the bio sorbents, viz. natural bamboo saw dust (Zhao et al. 2012a). Moreover, the result is in line with previous findings obtained with activated carbon prepared from Ceiba pentandra hulls (Rao et al. 2006), Van apple pulp (Depci et al. 2012) and manganese oxide-coated carbon nanotubes (Wang et al. 2007) in the removal of several metal cations. Moreover, at very high pH values, i.e. beyond pH 9.0, there could be a mixed effect of adsorption and co-precipitation of Cd(II) on the surface of solids resulting in complete removal of these ions, i.e. about 100 % uptake of Cd(II) by these solids (Lee et al. 2010, 2012).

Effect of pH in the percent removal of Cd(II) by using (a) AC(N) and modified AC(N), and (b) AC(R) and modified AC(R)
Kinetics of adsorption
The time dependence adsorption data was collected in order to discuss the kinetics of adsorption process. The initial Cd(II) concentration was kept at 10.0 mg/L and pH 5.0 as constant with operating temperature of 25 ± 1 °C. The results obtained were presented graphically in Fig. 6. It was noted that during the initial period of contact, a fast and rapid uptake of Cd(II) occurred onto these solids, and within 60 min of contact, most of the Cd(II) was adsorbed. These results were in agreement with previous findings in which different biosorbents or inorganic ion exchangers were employed (Benguell and Benaissa 2002; Mittal et al. 2010a; Mishra et al. 1998b; Tiwari et al. 1999). Furthermore, the kinetic modelling was performed using the time dependence sorption data.

Kinetic study in the removal of Cd(II) by (a) AC(N) and modified AC(N), and (b) AC(R) and modified AC(R)
Kinetic modelling
The time dependence data was modelled with the pseudo-first order (Eq. 1) and pseudo-second order (Eq. 2) equations to its linear form (Bhatnagar and Minocha 2009; Gupta et al. 2007):
where q e and q t (milligrams per gram) are the amount of Cd(II) sorbed at equilibrium and at time t (minutes), respectively. K f (1 per minute) and K s (gram per milligram per minute) are the pseudo-first-order and pseudo-second-order rate constants, respectively. Plots were drawn between log (q e − q t) vs. t and t/q t vs. t in order to fit the time dependence sorption data. The outcome of the linear least square fitting data was employed to estimate the K f and K s values along with the correlation coefficient R 2, which were then returned in Table 1. The scrutiny of Table 1 clearly demonstrated that the sorption kinetics well explained the pseudo-second-order rate law than the pseudo-first-order rate kinetics since reasonably high correlation coefficient was achieved for the least square fitting of the second-order rate equation. Moreover, the values of q e(exp), i.e. amount of Cd(II) sorbed at equilibrium, were perhaps close to the q e(cal) values optimized by the pseudo-second-order rate equation. The applicability of pseudo-second-order rate equation indicated that Cd(II) was sorbed predominantly by the strong chemical forces forming chemical bonds on the surface functional groups of solids (Ho and McKay 2000).
Effect of sorptive concentration
Furthermore, the sorption of Cd(II) was observed at various initial concentrations of Cd(II), i.e. varied from 1.0 to 20.0 mg/L, keeping the initial pH 5.0 and temperature 25 ± 1 °C as constant. Results obtained for percent uptake against the initial Cd(II) concentrations were shown graphically in Fig. 7. It was noted that by increasing the Cd(II) concentration from 1.0 to 20.0 mg/L, the percent uptake of Cd(II) decreased, respectively, from 65.71 % to 23.65 % for AC(N), 100.00 % to 38.42 % for MIAC(N), 100.00 % to 30.12 % for IIAC(N), 67.43 % to 28.15 % for AC(R), 100.00 % to 42.30 % for MIAC(R) and 100.00 % to 42.94 % for IIAC(R). These figures also indicated that manganese- or iron-modified solids possessed relatively higher sorption capacity at all studied Cd(II) concentrations with reference to their respective AC solids, which conferred its strong selectivity towards these modified solids.

Effect of initial sorptive concentration in the removal of Cd(II) by (a) AC(N) and modified AC(N), and (b) AC(R) and modified AC(R)
Equilibrium modelling
The equilibrium state sorption data obtained at different Cd(II) concentrations were further utilized in equilibrium modelling studies using Freundlich and Langmuir adsorption isotherms to its linear form (Mittal et al. 2009; Tiwari et al. 2007). The plots were drawn between log C e vs. log q e and C e vs. C e/q e, respectively, for the Freundlich and Langmuir adsorption isotherms. The terms C e and q e refer to the equilibrium bulk sorptive concentration and amount adsorbed at equilibrium, respectively. The data were fitted to their linear form, and the Freundlich constants 1/n (adsorption intensity) and K f (maximum adsorption capacity) and the Langmuir constants q max (maximum monolayer capacity) and b (the Langmuir rate constant) were estimated. The computed Freundlich and Langmuir constants along with the R 2 values were returned in Table 2.
Table 2 clearly indicated that both the adsorption isotherms were not fitted well for all the systems. However, the Freundlich model was relatively fitted well for these studied systems. The fractional values obtained for Freundlich constant 1/n (0 < 1/n < 1) indicated the heterogeneous surface structure of solids with an exponential distribution of active sites (Lee et al. 2010; Mishra et al. 1998a). Similarly, the Freundlich sorption capacity obtained is relatively higher and showed a strong affinity of the solids towards Cd(II). Moreover, the applicability of Freundlich equation pointed out that strong chemical forces were involved at the solid solution interface, and the sorbed ions were likely to be interacting laterally. On the other hand, the Langmuir constants were estimated for these systems, and the Langmuir monolayer capacity for these solids was obtained. Similarly, the lower values of Langmuir constant (b) reflected strong affinity of these solids towards Cd(II) (Gupta et al. 2005). Moreover, in general, it was found that the manganese- or iron-modified activated carbon solids possessed relatively higher removal capacities at least in the removal of Cd(II) from aqueous solutions.
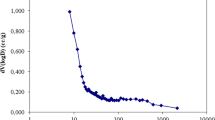
Effect of background electrolyte concentration
The background electrolyte concentration is an important parameter revealing the nature of binding of the sorptive ions onto the solid surface. It was demonstrated previously that the specific adsorption was not significantly affected with the change in background electrolyte concentrations, whereas the nonspecific adsorption was greatly affected with the change in background electrolyte concentration (Hayes et al. 1988). Therefore, keeping in view the foregoing discussion, the background electrolyte concentration dependence adsorption data was collected by increasing the NaNO3 concentrations from 0.001 to 1.0 mol/L. The initial Cd(II) concentration of 10.0 mg/L, pH 4.5 and temperature 25 ± 1 °C were kept constant. The results obtained were returned with percent removal versus background electrolyte concentration in Fig. 8. It was observed that 1,000 times increase in the background electrolyte concentration decreased the percent removal of Cd(II) only by 7.84 %, 6.34 %, 3.52 %, 4.93 %, 6.71 % and 10.07 %, respectively, for the solids AC(N), AC(R), MIAC(N), MIAC(R), IIAC(N) and IIAC(R). The insignificant decrease in percent removal of Cd(II) even by an increase of 1,000 times background electrolyte concentration strongly suggested that the Cd(II) ions were sorbed specifically onto the solid surface, forming predominantly inner sphere complexes (Payne and Abdel-Fattah 2005; Schmidt et al. 2008). Similar observations were reported previously where the copper ion was forming an inner sphere complex with sawdust; the study was supported reasonably well with theoretical modelling as well (Zhao et al. 2012b).

Effect of background electrolyte concentrations in the removal of Cd(II) by (a) AC(N) and modified AC(N), and (b) AC(R) and modified AC(R)
Column reactor operations
The breakthrough data was obtained by performing the column studies. The column conditions were maintained as influent concentration of Cd(II): 10.0 mg/L having a constant pH 5.0. The column was run as stated earlier, and the effluent samples were collected and subjected for bulk sorptive concentration. Furthermore, the breakthrough curves were shown in Fig. 9. The figures clearly indicated that a complete breakthrough for Cd(II) was obtained at the throughput volume of 0.30, 0.43, 0.39, 0.38, 0.42 and 0.38 L, respectively, for AC(N), MIAC(N), IIAC(N), AC(R), MIAC(R) and IIAC(R). These results inferred that these solid materials showed fairly good affinity towards the Cd(II) sorption. Hence, there could be an enhanced loading capacity of Cd(II) even under the dynamic conditions as well. The similar breakthrough data was obtained under the fixed-bed column operation in the decoloration of hazardous triarylmethane dye, Light Green SF (Yellowish), by employing the waste materials (Mittal et al. 2010b, c) metal cations by iron- or manganese-coated sand (Lee et al. 2012). The breakthrough data was further utilized to optimize the loading capacity of these solids under the dynamic conditions using the Thomas equation to its non-linear form (Thomas 1944):
where C e and C 0 are the Cd(II) concentration (milligrams per litre) of effluent and influent solutions, K T is the Thomas rate constant (litre per minute per milligram), q 0 the maximum amount of Cd(II) to be loaded (milligrams per gram) in the column under the specified column conditions, m is the mass of adsorbent packed in the column (gram), V is the throughput volume (litre) and Q is the flow rate of pumped Cd(II) solution (litres per minute). A non-linear regression was conducted using the column data by the least square fitting for two unknown parameters, i.e. K T and q 0 (cf. Fig. 9). The values obtained were returned in Table 3. The marked difference in loading capacity occurred for the modified activated carbon samples compared to the bare activated carbons, suggesting the comparable use of these modified solids in the selective removal of Cd(II) contaminated wastewater. These results were in line with the findings of batch reactor operations and previous reports in which Thomas equations were fitted well to demonstrate the loading capacity of different sorbents (Lee et al. 2012; Singh et al. 2012).

Breakthrough curves in the removal of Cd(II) by (a) AC(N) and modified AC(N), and (b) AC(R) and modified AC(R)
Conclusions
The activated carbon samples obtained precursor to different dead biomasses, viz. rice hulls and areca nut waste. The high specific surface area was utilized to immobilize the small-sized manganese or iron oxide onto the surface or within the pores of ACs. The materials were characterized with the IR and XRD data. XRD data showed that a characteristic peak of akaganeite (β-FeOOH) appeared with the IIAC samples. On the other hand, MIAC (rice hulls) showed the presence of Mn3O4 mineral phase of manganese. The surface morphology was obtained by the SEM images, which showed that iron or manganese oxide particles were clustered onto the solid surfaces. Furthermore, the solids were assessed for possible implication in attenuation of Cd(II) from aqueous solutions under the batch and reactor studies. Various parametric studies obtained by batch reactor operations enabled that increase in pH (2.0 to 10.0) favoured the percent uptake of Cd(II), whereas increase in concentration (1.0 to 20 mg/L) favoured the amount of Cd(II) removed. Interestingly, the increase in background electrolyte concentrations (0.01 to 1.0 mol/L NaNO3) was insignificantly changed in percent uptake of Cd(II) by these solids indicating that Cd(II) was forming strong chemical bonds with the available surface functional groups. The kinetic data fitted well to the pseudo-second-order rate kinetics, and the equilibrium state modelling showed the applicability of Freundlich adsorption isotherm. On the other hand, the breakthrough curves obtained by the column studies fitted well to the Thomas equation, and hence, the loading capacities of these solids were estimated which were found to be 1.524, 2.124, 2.265, 2.036, 2.197 and 2.723 mg/g, respectively, for the solids AC(N), MIAC(N), IIAC(N), AC(R), MIAC(R) and IIAC(R). The manganese- or iron-modified solids possessed somewhat higher removal capacity for Cd(II) hence could have enhanced application in the remediation of aquatic environment contaminated with Cd(II).
References
Al Bahri M, Calvo L, Gilarranz MA, Rodriguez JJ (2012) Activated carbon from grape seeds upon chemical activation with phosphoric acid: application to the adsorption of diuron from water. Chem Eng J 203:348–356
Amoyaw PA, Williams M, Bu XR (2009) The fast removal of low concentration of cadmium(II) from aqueous media by chelating polymers with salicylaldehyde units. J Hazard Mat 170:22–26
Barreira LD, Lito PF, Antunes B, Otero M, Lin Z, Rocha J, Pereira E, Duarte AC, Silva CM (2009) Effect of pH on cadmium(II) removal from aqueous solution using titanosilicate ETS-4. Chem Eng J 155:728–735
Benaissa H (2006) Screening of new sorbent materials for cadmium removal from aqueous solutions. J Hazard Mater B132:189–195
Benguell B, Benaissa H (2002) Cadmium removal from aqueous solution by chitin: kinetic and equilibrium studies. Wat Res 36:2463–2474
Berbenni P, Police A, Canziani R, Stabile L, Nobili F (2000) Removal of iron and manganese from hydrocarbon-contaminated groundwaters. Bioresource Technol 74:109–114
Bhatnagar A, Minocha AK (2009) Utilization of industrial waste for cadmium removal from water and immobilization in cement. Chem Eng J 150:145–151
Boparai HK, Joseph M, O'Carroll DM (2011) Kinetics and thermodynamics of cadmium ion removal by adsorption onto nano zerovalent iron particles. J Hazard Mater 186:458–465
Corapcioglu MO, Huang CP (1987) The adsorption of heavy metals onto activated carbon. Wat Res 21:1031–1044
Depci T (2012) Comparison of activated carbon and iron impregnated activated carbon derived from Golbasi lignite to remove cyanide from water. Chem Eng J 181–182:467–478
Depci T, Ku AR, Onal Y (2012) Competitive adsorption of lead and zinc from aqueous solution on activated carbon prepared from Van apple pulp: study in single- and multi-solute systems. Chem Eng J 200–202:224–236
Egan SK, Bogler PM, Carrington CD (2007) Update of US FDA's total diet study food list and diets. J Expo Sci Environ Epidermiol 17:573–582
Fierro V, Muniz G, Gonzalez-Sanchezc G, Ballinsab ML, Celzarda A (2009) Arsenic removal by iron-doped activated carbons prepared by ferric chloride forced hydrolysis. J Haz Mater 168:430–437
Grassi M, Kaykioglu G, Belgiorno V, Lofrano G (2012) Removal of emerging contaminants from water and wastewater by adsorption process. In: Lofrano G (ed) Emerging compounds removal from wastewater. Springer briefs in green chemistry for sustainability. doi:10.1007/978-94-007-3916-1_2
Goering PL, Waalkes MP, Klaassen CD (1994) Toxicology of cadmium. In: Goyer RA, Cherian MG (eds) Handbook of experimental pharmacology: toxicology of metals, biochemical effects, vol 115. Springer, New York, pp 189–214
GOI (Government of India) (2008) Ministry of Health and Family Welfare, Notification, GSR759 (E)
Gomez-Serrano V, Pastor-Villegas J, Perez-Florindo A, Duran-Valle C, Valenzuela-Calahorro C (1996) FT-IR study of rockrose and of char and activated carbon. J Anal Appl Pyrol 36:71–80
Gun'ko VM, Leboda R, Skubiszewska-Zieba J, Charmas PO (2005) Carbon adsorbents from waste ion-exchange resins. Carbon 43:1143–1150
Gupta VK, Saini VK, Jain N (2005) Adsorption of As(III) from aqueous solutions by iron oxide-coated sand. J Colloid Interface Sci 288:55–60
Gupta VK, Ali I, Saini VK (2007) Defluoridation of wastewaters using waste carbons slurry. Wat Res 41:3307–3316
Gupta VK, Mittal A, Malviya A, Mittal J (2009) Adsorption of carmoisine A from wastewater using waste materials—bottom ash and deoiled soya. J Colloid Interf Sci 335:24–33
Gupta VK, Gupta B, Rastogi A, Agarwal S, Nayak A (2011a) A comparative investigation on adsorption performances of mesoporous activated carbon prepared from waste rubber tire and activated carbon for a hazardous azo dye—Acid Blue 113. J Haz Mat 186:891–901
Gupta VK, Agarwal S, Saleh TA (2011b) Chromium removal by combining the magnetic properties of iron oxide with adsorption properties of carbon nanotubes. Wat Res 45:2207–2212
Hamdaoui O (2009) Removal of cadmium from aqueous medium under ultrasound assistance using olive leaves as sorbent. Chem Eng Processing 48:1157–1166
Hayes KF, Papelis C, Leckie JO (1988) Modeling ionic strength effects on anion adsorption at hydrous oxide/solution interfaces. J Colloid Interf Sci 125:717–726
Ho YS, McKay G (2001) The kinetics of sorption of divalent metal ions onto Sphagnum moss peat. Water Res 34:735–742
Hydari S, Sharififard H, Nabavinia M, Parvizi MR (2012) A comparative investigation on removal performances of commercial activated carbon, chitosan biosorbent and chitosan/activated carbon composite for cadmium. Chem Eng J 193–194:276–282
IARC (International Agency for Research on Cancer) (1993) Beryllium, cadmium, mercury and exposures in the glass manufacturing industry. IARC monographs on the evaluation of carcinogenic risk of chemicals to humans, Vol. 58. International Agency for Research on Cancer, Lyon France, pp 444. http://monographs.iarc.fr/ENG/Monographs/vol58/volume58.pdf
Irani M, Keshtkar AR, Moosaviaan MA (2012) Removal of cadmium from aqueous solution using mesoporous PVA/TEOS/APTES composite nanofiber prepared by sol-gel/electrospinning. Chem Eng J 200–202:192–201
Jarup L, Akesson A (2009) Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol 238:201–208
Katircioglu H, Aslim B, Turker AR, Atici T, Beyatli Y (2008) Removal of cadmium(II) ion from aqueous system by dry biomass, immobilized live and heat-inactivated Oscillatoria sp. H1 isolated from freshwater (Mogan Lake). Bioresource Technol 99:4185–4191
Klassen CD, Liu J, Choudhuri S (1999) Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol 39:267–294
Kong J, Yue Q, Huang L, Gao Y, Sun Y, Gao B, Li Q, Wang Y (2013) Preparation, characterization and evaluation of adsorptive properties of leather waste based activated carbon via physical and chemical activation. Chem Eng J. doi:10.1016/j.cej.2013.02.021
Larsen EH, Andersen NL, Moller A, Petersen A, Mortensen GK, Petersen J (2002) Monitoring the content and intake of trace elements from food in Denmark. Food Addit Contam 19:33–46
Lee SM, Kim WG, Laldawngliana C, Tiwari D (2010) Removal behavior of surface modified sand for Cd(II) and Cr(VI) from aqueous solutions. J Chem Eng Data 55:3089–3094
Lee SM, Laldawngliana C, Tiwari D (2012) Iron oxide nano-particles-immobilized material in the treatment of Cu(II), Cd(II) and Pb(II) contaminated waste waters. Chem Eng J 195–196:103–111
Li YS, Church JS, Woodhead AL, Moussa F (2010) Preparation and characterization of silica coated iron oxide magnetic nano-particles. Spectrochim Acta A 76:484–489
Liu Y, Lu X, Wu F, Deng N (2011) Adsorption and photooxidation of pharmaceuticals and personal care products on clay minerals. React Kinet Mech Catal 104:61–73
Liu Y, Guo Y, Gao W, Wang Z, Ma Y, Wang Z (2012) Simultaneous preparation of silica and activated carbon from rice husk ash. J Cleaner Production 32:204–209
Mishra SP, Tiwari D, Dubey RS, Mishra M (1998a) Biosorptive behavior of casein for Zn2+, Hg2+ and Cr3+: effects of physico-chemical treatments. Bioresource Technol 63:1–5
Mishra SP, Singh VK, Tiwari D (1998b) Radiotracer technique in adsorption study: part XVII. Removal behavior of alkali metal (K- and Li-) titanates for Cd(II). Appl Radiat Isot 49:1467–1475
Mittal A, Gupta VK, Malviya A, Mittal J (2008) Process development for the batch and bulk removal and recovery of a hazardous, water-soluble azo dye (Mentanil Yellow) by adsorption over waste materials (bottom ash and deoiled soya). J Haz Mat 151:821–832
Mittal A, Mittal J, Malviya A, Gupta VK (2009) Adsorptive removal of hazardous anionic dye “Congo red” from wastewater using waste materials and recovery by adsorption. J Colloid Interf Sci 340:16–26
Mittal A, Mittal J, Malviya A, Gupta VK (2010a) Removal and recovery of chrysoidine Y from aqueous solutions by waste materials. J Colloid Inetrf Sci 344:497–507
Mittal A, Mittal J, Malviya A, Kaur D, Gupta VK (2010b) Decoloration treatment of a hazardous triarylmethane dye, Light Green SF (Yellowish) by waste materials adsorbents. J Colloid Interf Sci 342:518–527
Mittal A, Mittal J, Malviya A, Kaur D, Gupta VK (2010c) Adsorption of hazardous dye crystal violet from wastewater by waste materials. J Colloid Interf Sci 343:463–473
Momcilovic M, Purenovic M, Bojic A, Zarubica A, Randelovic M (2011) Removal of lead (II) ions from aqueous solutions by adsorption onto pine cone activated carbon. Desalination 276:53–59
Mulligan CN, Yong RN, Gibbs BF (2001) Remediation technologies for metal-contaminated soils and groundwater. An evaluation Eng Geol 60:193–207
Muniz G, Fierro V, Celzard A, Furdin G, Gonzalez-Sanchez G, Ballinas ML (2009) Synthesis, characterization and performance in arsenic removal of iron-doped activated carbons prepared by impregnation with Fe(III) and Fe(II). J Haz Mat 165:893–902
Nakamoto K (1997) Infrared and raman spectra of inorganic and coordination compounds. Parts A and B. Wiley, New York
Olsson IM, Bensryd I, Lundh T, Ottosson H, Skerfving S, Oskarsson A (2002) Cadmium in blood and urine-impact of sex, age, dietary intake, iron status and former smoking-association of renal effects. Environ Health Perspect 110:1185–1190
Pandey PK, Verma Y, Choubey S, Pandey M, Chandrasekhar K (2008) Biosorptive removal of cadmium from contaminated groundwater and industrial effluents. Bioresour Technol 99:4420–4427
Payne KB, Abdel-Fattah TM (2005) Adsorption of arsenate and arsenite by iron-treated activated carbon and zeolites: effects of pH, temperature and ionic strength. J Environ Sci Health 40:723–774
Perez-Rama M, Alonso JA, Lopez CH, Vaamonde ET (2002) Cadmium removal by living cells of the marine microalga Tetraselmis suecica. Bioresource Technol 84:265–270
Puziy AM, Poddubnaya OI, Martinez-Alonso A, Suarez-Garcia F, Tascon JMD (2002) Synthetic carbons activated with phosphoric acid I. Surface chemistry and ion binding properties. Carbon 40:1493–1505
Rao MM, Ramesh A, Rao GPC, Seshaiah K (2006) Removal of copper and cadmium from the aqueous solutions by activated carbon derived from Ceiba pentandra hulls. J Hazard Mat B129:123–129
Rodulfo-Baechler SM, Gonalez-Cortes SL, Orozco J, Sagredo V, Fontal B, Mora AJ, Delgado G (2004) Characterization of modified iron catalysts by X-ray diffraction, infrared spectroscopy, magnetic susceptibility and thermo gravimetric analysis. Mater Lett 58:2447–2450
Salim R, Al-Subu M, Dawod E (2008) Efficiency of removal of cadmium from aqueous solutions by plant leaves and the effects of interaction of combinations of leaves on their removal efficiency. J Environ Management 87:521–532
Schmidt TD, Vlasova N, Zuzaan D, Kersten M, Daus B (2008) Adsorption mechanism of arsenate by zirconyl-functionalized activated carbon. J Colloid Interface Sci 317:228–234
Schutte R, Nawrot TS, Richart T, Thijs L, Vavderschueren D, Kuznetsova T, Van Hecke E, Roels HA, Staessen JA (2008) Bone resorption and environmental exposure to cadmium in women: a population study. Environ Health Perspect 116:777–783
Sharma YC (2008) Thermodynamics of removal of cadmium by adsorption on an indigenous clay. Chem Eng J 145:64–68
Si H, Wang H, Shen H, Zhou C, Li S, Lou S, Xu W, Du Z, Song L (2009) Controlled synthesis of monodisperse manganese oxide nanocrystals. Cryst Eng Comm 11:1128–1132
Singh A, Kumar D, Gaur JP (2012) Continuous metal removal from solution and industrial effluents using Spirogyra biomass-packed column reactor. Wat Res 46:779–788
Tiwari D, Mishra SP, Mishra M, Dubey RS (1999) Biosorptive behavior of Mango (Mangifera indica) and Neem (Azadirachta indica) bark for Hg2+, Cr3+ and Cd2+ toxic ions from aqueous solutions: a radiotracer study. Appl Radiat Isot 50:631–642
Tiwari D, Kim HU, Lee SM (2007) Removal behavior of sericite for Cu(II) and Pb(II) from aqueous solutions: batch and column studies. Sep Purif Technol 57:11–16
Tiwari D, Laldanwngliana C, Choi CH, Lee SM (2011) Manganese-modified natural sand in the remediation of aquatic environment contaminated with heavy metal toxic ions. Chem Eng J 171:958–966
Thomas HC (1944) Heterogeneous ion exchange in a flowing system. J Am Chem Soc 66:1664–1666
Vilensky MY, Berkowitz B, Warshawsky A (2002) In situ remediation of groundwater contaminated by heavy- and transition-metal ions by ion-exchange methods. Environ Sci Technol 36:1851–1855
Vitela-Rodriguez AV, Rangel-Mendez JR (2013) Arsenic removal by modified activated carbons with iron hydro(oxide) nanoparticles. J Environ Management 114:225–231
Waalkes MP (2000) Cadmium carcinogenesis in review. J Inorg Biochem 79:241–244
Waalkes MP (2003) Cadmium carcinogenesis. Mutation Res 533:107–120
Waalkes MP, Misra RR (1996) Cadmium carcinogenicity and genotoxicity. In: Chang L (ed) Toxicology of metals. CRC, Boca Raton, pp 231–244
Waalkes MP, Anyer M, Diwan B (1999) Chronic toxic and carcinogenic effects of oral cadmium in the noble (NBL/Cr) rat: induction of neoplastic and proliferative lesions of the adrenal, kidney, prostate and testes. J Toxicol Environ Health 58:199–214
Wang SG, Gong WX, Liu XW, Yao YW, Gao BY, Yue QY (2007) Removal of lead(II) from aqueous solution by adsorption onto manganese oxide-coated carbon nanotubes. Sep Purif Technol 58:17–23
Wang HQ, Yang GF, Li QY, Zhong XX, Wang FP, Li ZS, Li YH (2011) Porous nano- MnO2: large scale synthesis via a facile quick-redox procedure and application in a supercapacitor. New J Chem 35:469–475
WHO (2008) Guidelines for drinking water quality: recommendations, vol 1, 3rd edn. World Health Organization, Geneva
Xu J, Wu L, Chang AC (2009) Degradation and adsorption of selected pharmaceuticals and personal care products (PPCPs) in agricultural soils. Chemosphere 77:1299–1305
Yin P, Yu Q, Jin B, Ling Z (1999) Biosorption removal of cadmium from aqueous solution by using pretreated fungal biomass cultured from starch wastewater. Wat Res 33:1960–1963
Zhao XT, Zeng T, Hu ZJ, Gao HW, Zou CY (2012a) Modeling and mechanism of the adsorption of proton onto natural bamboo sawdust. Carbohydr Polym 87:1199–1205
Zhao XT, Zeng T, Li XY, Hu ZJ, Gao HW, Xie Z (2012b) Modified and mechanism of the adsorption of copper ion onto natural bamboo sawdust. Carbohydr Polym 89:185–192
Zhu ZL, Ma HM, Zhang RH, Ge YX, Zhao JF (2007) Removal of cadmium using MnO2 loaded D301 resin. J Environ Sci 19:652–656
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST; No. 2012R1A2A4A01001539).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Vinod Kumar Gupta
Rights and permissions
About this article
Cite this article
Lee, SM., Lalhmunsiama, Choi, SI. et al. Manganese and iron oxide immobilized activated carbons precursor to dead biomasses in the remediation of cadmium-contaminated waters. Environ Sci Pollut Res 20, 7464–7477 (2013). https://doi.org/10.1007/s11356-013-1609-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-013-1609-x