
Abstract
Functionalized magnetic core–zeolitic shell nanocomposites were prepared via hydrothermal and precipitation methods. The products were characterized by vibrating sample magnetometer, X-ray powder diffraction, Fourier transform infrared spectroscopy, nitrogen adsorption–desorption isotherms, and transmission electron microscopy analysis. The growth of mordenite nanocrystals on the outer surface of silica-coated magnetic nanoparticles at the presence of organic templates was well approved. The removal performance and the selectivity of mixed metal ions (Pb2+ and Cd2+) in aqueous solution were investigated via the sorption process. The batch method was employed to study the sorption kinetic, sorption isotherms, and pH effect. The removal mechanism of metal ions was done by chem–phys sorption and ion exchange processes through the zeolitic channels and pores. The experimental data were well fitted by the appropriate kinetic models. The sorption rate and sorption capacity of metal ions could be significantly improved by optimizing the parameter values.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Elements having atomic weight between 63.5 and 200.6, and a specific gravity greater than 5.0 are known as heavy metals (Srivastava and Majumder 2008). While some of their ions are known to be toxic or carcinogenic, these metals are not biodegradable and tend to accumulate in living organisms. Toxic heavy metals of particular concern in treatment of industrial wastewaters include zinc, copper, nickel, mercury, cadmium, lead, and chromium (Fu and Wang 2011). Cadmium, zinc, copper, nickel, lead, mercury, and chromium are often detected in industrial wastewaters, which originate from metal plating, mining activities, smelting, battery manufacture, tanneries, petroleum refining, paint manufacture, pesticides, pigment manufacture, printing and photographic industries, etc., (Ngah and Hanafiah 2008; Jain et al. 1997; Srivastava et al. 1995; Gupta et al. 2009a, Gupta et al. 2007a; Goyal et al. 2007). Heavy metal ions and the other impurities in wastewater can be removed from polluted waters using a wide range of methods such as solvent extraction, precipitation, vacuum evaporation, membrane technologies, ionic exchange, and adsorption (Gupta et al., 2000, 2006a, 2006b, 2007b, c, d, e, 2009b, 2010; Gupta and Rastogi 2009; Gupta and Sharma 2003; Ali 2010, 2012; Ali et al. 2012; Mittal et al. 2005; Jain et al. 2004; Kim et al. 2000; Alvarez-Ayuso and Garcia-Sanchez 2003; Alvarez-Ayuso et al. 2003; Erdem et al. 2004).
Over the past decades, zeolites have a large applicability for decontamination, purification of urban and industrial residual waters, protection of waste disposal areas, purification of industrial gases, etc. Many researchers studied using natural and synthetic zeolites to remove heavy metal ions in the aqueous medium (Langella et al. 2000; Cincotti et al. 2001; Badillo-Almaraz et al. 2003; Peric et al. 2004). Properties such as high surface area, individual micro-pores, a variety of channels, and high resistance make them very useful for industrial applications and academic research.
The combination of zeolitic materials with magnetic and/or active functional groups to form core–shell structured composite is undoubtedly of special interest in diagnostic analysis (Levy et al. 2002), bioseparation (Li et al. 2007), and controlled drug release (Murray et al. 1993; Yang et al. 2008; Arruebo et al. 2006) based on their unique magnetic responsivity, low cytotoxicity, good biocompatibility, and mesoporous properties. Over the past decade, the preparation of multifunctional microspheres consisting of a magnetite core with a mesoporous shell has been reported (Giri et al. 2005; Zhao et al. 2005; Deng et al. 2008; Guo et al. 2006; Kim et al. 2006). The uniform-sized magnetic particles were normally prepared via a high-temperature decomposition method (Kim et al. 2006), or hydrothermal process (Deng et al. 2008). The formation of the core–shell nanocomposites is conventionally followed by an encapsulation procedure, where the magnetite core is encapsulated by a silica layer using a sol–gel technique (Piaoping et al. 2009).
The aim of the present study was to investigate the heavy metal ions sorption characteristics of structured magnetic core–zeolitic shell nanocomposites. The mechanism of heavy metals sorption over the different composites, sorption kinetic, and sorption isotherms was discussed. The dependence of sorption rate to the pH of solution was studied too.
Experimental
Materials, instruments, and methods
Reagent grade chemicals such as NaOH, tetramethylammoniumhydroxide (TMAOH) (10 % w/w), ethylene glycol (EG), cyclohexane, cyclohexanole, C14H22O(C2H4O) n (Triton X-100) 100, 3-glycidoxypropyltrimethoxysilane (GPTS), glutamic acid (GLU) and various salts of polyvalent metals were used as required. Tetraethylorthosilicate (TEOS) was used as the silica source. All chemicals were purchased from Merck chemical Co. (Germany) and were used without any further purification. Water samples contaminated with heavy metals were prepared using their respective metal ion salts.
The X-ray diffraction (XRD) patterns of the prepared samples were recorded on a Bruker D8 advance X-ray diffractometer with CuKα irradiation (λ = 0.15406 nm). The Fourier transform infrared spectroscopy (FTIR) spectra were recorded using the NB series spectrometer. The specific surface area of the nanocomposites was calculated from the N2 adsorption–desorption isotherm at 77 K, using Belsorp apparatus (Japan). The average particle size and morphology of the samples were examined by transmission electron microscope (TEM). Magnetic studies were carried out on a TOEI VSM-5 vibrating sample magnetometer (VSM) at 300 K.
Core–shell nanocomposites preparation
Synthesis of magnetic nanoparticles (NiFe2O4)
Magnetic nanoparticles were prepared via hydrothermal method in a 200-ml stainless steel autoclave with a Teflon liner under autogenous pressure. In a typical procedure, a 50-ml transparent solution containing Ni(NO3)2 and FeCl3 (corresponding to Ni2+/Fe3+ molar ratio of 1:2 ) was prepared and added to 50 ml of NaOH solution 2 M dropwise under magnetic stirring. Then a mixture includes EG and TMAOH was added to the above suspension dropwise. After stirring for 2 h, the resultant mixture was immediately transferred into the autoclave and kept at 200 °C for 8 h. After this time, the resulting solid products were collected by filtration, repeatedly washed with double-distilled water, and then dried 80 °C for 6 h.
Silica coating of magnetic nanoparticles
Silica-coated NiFe2O4 nanoparticles were prepared by water-in-oil microemulsion approach (Liu et al. 2010) with some modifications. Cyclohexane (120 mL), cyclohexanol (30 mL) and Triton X-100 (30 mL) were placed in a round-bottom flask and the solution was stirred. Once the mixture was homogeneous, an aqueous suspension of NiFe2O4 nanoparticles (5 mL) was added to the above solution. The suspension was continuously stirred for 1 h. Ammonium (4 mL) and TEOS (4 mL) were then added. After the mixture was stirred at room temperature for 24 h, the reaction solution was decanted with the aid of the magnet. The obtained silica-coated nanoparticles were redispersed several times in a mixture of double-distilled water and ethanol, separated magnetically, and dried at 80 °C for 12 h.
Growth of mordenite nanocrystals on the SiO2@NiFe2O4 surface
Mordenite nanocrystals were grown on the surface of primary synthesized cores (SiO2@NiFe2O4) by hydrothermal treatment and at the presence of organic templates. In a typical procedure, Al(NO3)3·9H2O is dissolved in alkaline solution of NaOH 6 M. TMAOH and EG were added to the prepared solution under stirring. TEOS was added dropwise to the above solution to obtain a gel where then magnetically stirred for 3 h on a magnetic stirrer. The reluctant mixture was immediately placed in a 200-ml autoclave which was then maintained in a preheated oven at autogenous pressure and static conditions. After the completion of the period of synthesis (24 h and 180 °C), the product was filtered and washed repeatedly with double distilled water and dried at 90 °C for 10 h. Finally, the solid powder was calcined at 500 °C for 5 h and denoted Z@SiO2@NiFe2O4.
Surface modification with GPTS–GLU
Z@SiO2@NiFe2O4 nanocomposites were modified with GPTS and GLU according to the earlier work with some modifications (Zhiya et al. 2006). GLU was dissolved in double-distilled water, and the obtained suspension was adjusted to pH 11 with NaOH 10 M. The solution was transferred into a flask bottle placed in the ice-bath at 0 °C, and GPTS was slowly added while being stirred. The mixed solution was heated to 65 °C for 2 h by stirring. After adjusting the pH of the prepared solution to 6 with concentrated HCl, an aqueous suspension of Z@SiO2@NiFe2O4 was added and stirred for 12 h. The resulting product was separated with the help of the permanent magnet, washed thoroughly with distilled water, dried at 80 °C for 6 h and denoted GPTS–GLU@Z@SiO2@NiFe2O4.
Sorption test
All experiments were carried out in a 1-L batch reactor with the initial X(II) concentration (X = Pb and Cd) of 20 mg/L at the initial pH value 5. The sorbent mass was fixed at 0.1 g. The reactor was stirred with a magnetic stirrer operated at 300 rpm. At predetermined time intervals, 3 mL samples were taken from the reactor, centrifuged, and residual X(II) concentration was measured with an atomic absorption spectrophotometer. By performing appropriate material balance, the quantity of X(II) adsorbed at the selected time intervals was determined and used for kinetic analysis.
Results and discussion
Characterization
VSM analysis
The magnetic properties of the core–shell nanocomposites were characterized and the results are shown in Fig. 1. Magnetic measurement shows that pure NiFe2O4, SiO2@NiFe2O4, Z@SiO2@NiFe2O4, and GPTS–GLU@Z@SiO2@NiFe2O4 have magnetic saturation values of 48, 36.1, 15, and 12.5 emu/g, respectively. It is important that the growth of the zeolitic layer on the surface decrease the magnetic saturation to less than half. It should also be noted that the multiple modified NiFe2O4 cores still show good magnetization, indicating their suitability for using and separation. The magnified hysteresis loops in Fig. 1 confirm the superparamagnetic feature for all the samples. Moreover, the multifunctional core–shell nanocomposites with homogenous dispersion exhibit the re-disperse properties and a suitable response to the external magnetic field due to its high magnetization.

Magnetic saturation results: a NiFe2O4, b SiO2@NiFe2O4, c Z@SiO2@NiFe2O4, d GPTS–GLU@Z@SiO2@NiFe2O4
XRD analysis
XRD patterns of synthesized magnetic core–zeolitic shell nanocomposites are presented in Fig. 2. Diffraction characteristic peaks of NiFe2O4 nanoparticles and MOR-type zeolite were seen in the samples. The strong peaks in 2θ = 30°, 35.5°, 43°, 57.2°, and 63° are attributed to NiFe2O4 nanoparticles. The zeolitic phases were identified by comparing the sharp diffraction peaks with the data reported in International Zeolite Association. The characteristic peaks for mordenite nanocrystals are well recognizable by their green colors. GPTS–GLU@Z@SiO2@NiFe2O4 and Z@SiO2@NiFe2O4 almost have the same XRD peaks, too and no excess crystalline phase appears after the surface modification.

The XRD patterns of prepared samples: a NiFe2O4, b Z@SiO2@NiFe2O4, and c GPTS–GLU@Z@SiO2@NiFe2O4
FTIR analysis
The FTIR spectra of NiFe2O4 magnetic nanoparticles and synthesized core–shell nanocomposites are shown in Fig. 3. In the FTIR spectrum of pure magnetic nanoparticles and modified nanocomposites, the strong band of OH (in higher than 3,400 cm−1) and a characteristic peak of H2O in 1,630 cm−1 suggest that a large number of OH groups and H2O molecules, which exist on the surface, play a key role in adsorbing heavy metal ions and the other impurities in wastewater (Lu et al. 2010; Jal et al. 2004). The absorption band related to Si–O–Si (in 1,092 cm−1) also appears in the spectrum of SiO2@NiFe2O4. All bands observed in the ranges of 920–1,250, 650–720, and 420–500 cm−1 in the Z@SiO2@NiFe2O4 spectrum correspond to the internal tetrahedral: asymmetrical stretch, symmetrical stretch, and T–O bonds (where T = Si or Al in the zeolitic structure), respectively. The peaks below 550 cm−1 are generally attributed to δ (O–T–O) bending and rotation modes. Some peaks between 700 and 850 cm−1 and between 1,000 and 1,150 cm−1 are assigned to symmetric and antisymmetric T–O–T stretching vibrations. In contrast, the stretching vibrational mode for AlO−4 is located in the range 650–900 cm−1 (Sharma et al. 2008).

The FTIR spectrum of: a NiFe2O4, b SiO2@NiFe2O4, c Z@SiO2@NiFe2O4, and d GPTS–GLU@Z@SiO2@NiFe2O4
The absorption band related with X–O (X = Fe, Mn, Co, Ni) (579 cm−1) is also present in all spectrums.
BET analysis
The results for Brunauer–Emmett–Teller (BET) analysis of prepared samples are shown in Fig. 4 and Table 1. Figure 4 presents a typical hysteresis loop obtained from N2 adsorption–desorption procedure for sample Z@SiO2@NiFe2O4 and shows that the core–shell nanocomposites have a good porosity. The results revealed that the growth of the zeolitic layer on the outer surface of the magnetic core increases the specific surface area and improves the structure porosity. As shown, deposition of the GPTS–GLU layer on the surface has not changed the basic pore structure of the Z@SiO2@NiFe2O4 sample. However, the BET surface area and total pore volume decreased from 215.7 m2 g−1 and 0.16 cm3 g−1 to 201.58 m2 g−1 and 0.15 cm3 g−1, respectively. Pore size distributions of the prepared samples were determined by Barrett–Joyner–Halenda (BJH) method. BJH analysis can also be employed to determine the pore area and the specific pore volume using adsorption and desorption techniques. This technique characterizes pore size distribution independent of the external area due to the particle size of the sample. A typical BJH plot for Z@SiO2@NiFe2O4 is shown in Fig. 5. Figure 5 shows that the pore diameter ranges from 1 to 100 nm. As seen in this figure, most pore diameters are concentrated between 2–50 nm indicating the present mesopores.

N2 adsorption–desorption plot for Z@SiO2@NiFe2O4

BJH plot for Z@SiO2@NiFe2O4
TEM analysis
The morphological and structural features of the samples were further examined by TEM analysis as shown in Fig. 6. It is clear that the Z@SiO2@NiFe2O4 core–shell nanocomposite was of well spherical structure and high monodispersity in size. The average diameters were about 15–30 and 80–100 nm for the NiFe2O4 nanoparticles and Z@SiO2@NiFe2O4 nanocomposites, respectively. Upon deposition of the zeolitic shell to sample SiO2@NiFe2O4, the diameter of the particles increased significantly, demonstrating that the mordenite shell was ∼40 nm thick.

TEM images of: a synthesized magnetic nanoparticles and b core–shell nanostructures
Sorption of heavy metal ions over the different composites
Figure 7 shows the sorption curves of Pb2+ and Cd2+ over the prepared nanocomposites. While the removal efficiency is less over the SiO2@NiFe2O4 sample, the results showed that the growth of the zeolitic layer on the SiO2@NiFe2O4 core increased it. That was expected because of high porosity and the specific surface area of synthetic zeolitic materials. Furthermore, the metal ions in zeolitic channels could be exchanged with the present ions in the solution medium. When GPTS–GLU@Z@SiO2@NiFe2O4 is used as sorbent, in addition to the mentioned statements, heavy metal ions could be chemically sorbed by surrounding the X 2+ ions with carboxylate groups of GLU on the surface. It can be observed in all cases that the removal efficiency of Cd2+ is higher than Pb2+ ions. The radius size of Cd2+ is less than Pb2+ which makes it more suitable for effective ion exchange and also more effective bonding with organic groups on the surface. However, the presence of the GPTS–GLU organic layer on the surface can block some zeolitic pores and channels. The physisorption of next layers of metal ions on the surface can be possible. Thus, the highest removal is achieved with these nanocomposites. Scheme 1 explained the total removal process of heavy metal ions.

Removal efficiency over: a Z@SiO2@NiFe2O4, b GPTS–GLU@Z@SiO2@NiFe2O4, and c SiO2@NiFe2O4 (square: Cd2+ and diamond: Pb2+)

Total removal process of heavy metal ions
Sorption kinetic
Figure 8 shows the kinetic sorption of metal ions onto prepared nanocomposites. After fitting into the pseudo-first-order and pseudo-second-order sorption kinetic models, it was observed that pseudo-second-order model fitted well for both Pb2+ ion and Cd2+ ions and gave the best description of experimental data points. It is important that this model be based on the assumption that the rate limiting step may be a chemical sorption involving valence forces through sharing or electronic interaction between the adsorbent and the adsorbate (Taty-Costodes et al. 2003). The pseudo-second-order chemisorption kinetics rate equation is expressed as:
where q e and q t are the amounts of metal ions adsorbed onto the adsorbents (milligram per gram) at equilibrium and at time t, respectively. k 2 is the rate constant of second order (gram per milligram per minute).

Pseudo-second order kinetic plots for X(II) sorption onto a: Z@SiO2@NiFe2O4, b: GPTS–GLU@Z@SiO2@NiFe2O4, and c: SiO2@NiFe2O4 (square: Cd2+ and diamond: Pb2+)
The integrated form of Eq. 1 is rearranged and becomes:
in which the constants can be determined by plotting t/q t versus t.
Sorption isotherms
The development of an equation which accurately represents the results and could be used for design purposes is achieved by the analysis of the isotherm data. In order to investigate the sorption isotherm, two equilibrium models were applied: Langmuir and Freundlich isotherm equations. The first state is obtained under the ideal assumption of a totally homogeneous adsorption surface, whereas the second isotherm is appropriate for a highly heterogeneous surface. The Langmuir sorption isotherm is the best known of all isotherms describing sorption (Langmuir 1918) and it has been successfully applied to many sorption processes (Alvarez-Ayuso et al. 2003, Reddad et al. 2002, Namasivayam and Yamuna 1999, Beyazit et al. 2003; Hui et al. 2005). It is represented as:
Where C e is the equilibrium aqueous metal ions concentration (milligram per liter), q e the amount of metal ions adsorbed per gram of adsorbent at equilibrium (milligram per gram), q m and b are the Langmuir constants related to the maximum adsorption capacity and energy of adsorption, respectively. The values of q m (milligram per gram) and b (per milligram) can be determined from the linear plot of C e /q e versus C e . The Freundlich isotherm fairly satisfactory empirical isotherm can be used for a non-ideal sorption that involves heterogeneous sorption is most frequently used to describe the adsorption of inorganic and organic components in solution (Namasivayam et al. 1994, 2003), and is expressed as:
where K is roughly an indicator of the adsorption capacity and 1/n the adsorption intensity. By plotting log qe versus logCe, values of K and n can be determined from the slope and intercept of the plot.
For our experiments, the sorption data were fitted into the linearized Langmuir equation and the results are shown in Fig. 9 and Table 2.

Linearized Langmuir isotherm (Eq. 3) for Cd(II) ions sorption onto GPTS–GLU@Zeolite@SiO2@NiFe2O4. T = 25 °C
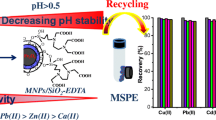
pH dependence of sorption efficiency
As shown in Fig. 10, the removal efficiency of metal ions was generally enhanced when the initial pH was increased. The pH of the aqueous solution is an important operational parameter in the adsorption process because it affects the solubility of the metal ions, concentration of the counter ions on the functional groups of the adsorbent and the degree of ionization of the adsorbate during reaction. The active sites on an adsorbent can either be protonated or deprotonated depending on the pH while at the same time the adsorbate speciation in a solution depends on the pH too. Lead for example, exists as Pb2+, PbOH+, and Pb(OH)− 3 depending on pH. This was due to two reasons: zeolites are highly selective for H3O+ ions when their concentration is high. Thus, at lower pH values the H3O+ ions compete with the metal ions for exchanging in zeolitic pores and channels (Shriver et al. 1990). Moreover, the functional groups on the zeolitic surface (two carboxylate group) could remain more anionic at higher pH that may make a significant contribution to the metal removal via chemsorption. Besides, the zeolitic surface is positively charged when the solution pH is lower. Thus the decrease in removal efficiency as the pH decreases can be attributed to the increase in positive charge on the zeolitic surface.

Effect of pH on the sorption of heavy metal ions onto GPTS–GLU@Zeolite@SiO2@NiFe2O4
Conclusions
Magnetic core–zeolitic shell nanocomposites were prepared by hydrothermal and coprecipitation methods. The analysis of the products well confirmed the growth of the mordenite layer on the surface of SiO2@NiFe2O4 nanoparticles which was performed by a new procedure. The VSM analysis method showed that the magnetic nanoparticles have a good magnetization which decreased by adding the layers on the surface. Prepared composites were applied for the removal of heavy metal ions Pb2+ and Cd2+ from waste water. The results showed that the surface modification influences strongly the removal efficiency. Studying the sorption kinetic and sorption isotherms indicated that the removal obeys the pseudo-second-order kinetic and Langmuir models, respectively. Finally, the effect of pH was investigated and according to the results, increasing the medium acidity increases the removal efficiency which is explainable with a glance at the ion exchange capacity and surface components. Finally, one can conclude that such zeolitic nanomaterials with a high specific surface area includes a magnetic core which could be applied for impressive removal of toxic species from waste water, while these materials can be separated from the medium by an external magnetic field and reused repeatedly. In addition, suitable surface modification provided an improved capacity for chemical sorption.
References
Ali I (2010) The quest for active carbon adsorbent substitutes: inexpensive adsorbents for toxic metal ions removal from wastewater. Sepn Purfn Rev 39:95–171
Ali I (2012) New generation adsorbents for water treatment. Chem Revs 112:5073–5091
Ali I, Asim M, Khan TA (2012) Low cost adsorbents for the removal of organic pollutants from wastewater. J Environ Manag 113:170–183
Alvarez-Ayuso E, Garcia-Sanchez A (2003) Removal of heavy metals from waste waters by natural and Na-exchanged bentonites. Clays Clay Miner 51:475–480
Alvarez-Ayuso E, Garcia-Sanchez A, Querol X (2003) Purification of metal electroplating waste waters using zeolites. Water Res 37:4855–4862
Arruebo M, Galan M, Navascues N, Tellez C, Marquina C, Ibarra MR (2006) Development of magnetic nanostructured silica-based materials as potential vectors for drug-delivery applications. Chem Mater 18:1911–1919
Badillo-Almaraz V, Trocellier P, Davila-Rangel I (2003) Adsorption of aqueous Zn(II) species on synthetic zeolites. Nucl Instrum Meth B 210:424–428
Beyazit N, Ergun QN, Peker I (2003) Cu(II) removal from aqueous solution using Dogantepe (Amasya) zeolites. Int J Environ Pollut 19:150–159
Cincotti A, Lai N, Orru R, Cao G (2001) Sardinian natural clinoptilolite for heavy metals and ammonium removal: experimental and modeling. Chem Eng J 84:275–282
Deng YH, Qi DW, Deng CH, Zhang XM, Zhao DY (2008) Superparamagnetic highmagnetization micropheres with a Fe3O4@SiO2 core and perpendicularly aligned mesoporous SiO2 shell for removal of microcystins. J Am Chem Soc 130:28–29
Erdem E, Karapinar N, Donat R (2004) The removal of heavy metal cations by natural zeolites. J Colloid Interface Sci 280:309–314
Fu F, Wang Q (2011) Removal of heavy metal ions from wastewaters: a review. J Environ Manage 92:407–418
Giri S, Trewyn BG, Stellmaker MP, Lin VSY (2005) Stimuli-responsive controlledrelease delivery system based on mesoporous silica nanorods capped with magnetic nanoparticles. Angew Chem Int Ed 44:5038–5044
Goyal RN, Gupta VK, Oyama M, Bachheti N (2007) Voltammetric determination of adenosine and guanosine using fullerene-C60-modified glassy carbon electrode. Talanta 71:1110–1117
Guo J, Yang WL, Wang CC, He J, Chen JY (2006) Poly(N-isopropylacrylamide)-coated luminescent/magnetic silica microspheres: preparation, characterization, and biomedical applications. Chem Mater 18:5554–5562
Gupta VK, Rastogi A (2009) Biosorption of hexavalent chromium by raw and acid-treated green alga Oedogonium hatei from aqueous solutions. J Hazard Mater 163:396–402
Gupta VK, Sharma S (2003) Removal of Zinc from Aqueous Solutions Using Bagasse Fly Ash − a Low Cost Adsorbent. Ind Eng Chem Res 42:6619–6624
Gupta VK, Srivastava SK, Tyagi R (2000) Design parameters for the treatment of phenolic wastes by carbon columns (obtained from fertilizer waste material). Water Res 34:1543–1550
Gupta VK, Mittal A, Krishnan L, Mittal J (2006a) Adsorption treatment and recovery of the hazardous dye, Brilliant Blue FCF, over bottom ash and de-oiled soya. J Colloid Interface Sci 293:16–26
Gupta VK, Mitall A, Kurup L, Mittal J (2006b) Adsorption of a hazardous dye, erythrosine, over hen feathers. J Colloid Interface Sci 304:52–57
Gupta VK, Jain R, Mittal A, Mathur M, Sikarwar S (2007a) Photochemical degradation of the hazardous dye Safranin-T using TiO2 catalyst. J Colloid Interface Sci 309:464–469
Gupta VK, Jain R, Varshney S (2007b) Removal of Reactofix golden yellow 3 RFN from aqueous solution using wheat husk—An agricultural waste. J Hazard Mater 142:443–448
Gupta VK, Singh AK, Gupta B (2007c) Schiff bases as cadmium(II) selective ionophores in polymeric membrane electrodes. Anal Chim Acta 583:340–348
Gupta VK, Ali I, Saini VK (2007d) Defluoridation of wastewaters using waste carbon slurry. Water Res 41:3307–3316
Gupta VK, Jain R, Varshney S (2007e) Electrochemical removal of the hazardous dye Reactofix Red 3 BFN from industrial effluents. J Colloid Interface Sci 312:292–296
Gupta VK, Khayat MA, Singh AK, Pal MK (2009a) Nano level detection of Cd(II) using poly(vinyl chloride) based membranes of Schiff bases. Anal Chim Acta 634:36–43
Gupta VK, Goyal RN, Sharma RA (2009b) Novel PVC Membrane Based Alizarin Sensor and its application; Determination of Vanadium, Zirconium and Molybdenum. Int J Electrochem Sci 4:156–172
Gupta VK, Rastogi A, Nayak A (2010) Adsorption studies on the removal of hexavalent chromium from aqueous solution using a low cost fertilizer industry waste material. J Colloid Interface Sci 342:135–141
Hui KS, Chao CYH, Kot SC (2005) Removal of mixed heavy metal ions in wastewater by zeolite 4A and residual products from recycled coal fly ash. J Hazard Mater 127:89–101
Jain AK, Gupta VK, Khurana U, Singh LP (1997) A new membrane sensor for UO ions based on 2-hydroxyacetophenoneoxime-thiourea-trioxane resin. Electroanalysis 9:857–860
Jain AK, Gupta VK, Jain S (2004) Removal of Chlorophenols Using Industrial Wastes. Environ Sci Technol 38:1195–1200
Jal PK, Patel S, Mishra BK (2004) Chemical modification of silica surface by immobilization of functional groups for extractive concentration of metal ions. Talanta 62:1005–1028
Kim JS, Park JC, Yi J (2000) Zinc ion removal from aqueous solutions using modified silica impregnated with 2-ethylhexyl 2-ethylhexyl phosphoric acid. Sep Sci Technol 35:1901–1916
Kim J, Lee JE, Lee J, Yu JH, Kim BC, An K (2006) Magnetic fluorescent delivery vehicle using uniform mesoporous silica spheres embedded with monodisperse magnetic and semiconductor nanocrystals. J Am Chem Soc 128:688–689
Langella A, Pansini M, Cappelletti P, Gennaro B, Gennaro M, Colella C (2000) NH4+, Cu2+, Zn2+, Cd2+ and Pb2+ exchange for Na+ in a sedimentary clinoptilolite, North Sardinia, Italy. Microporous and Mesoporous Mater 37:337–343
Langmuir I (1918) Adsorption of gases on plain surface of glass mica platinum. J Am Chem Soc 40:1361–1403
Levy L, Sahoo Y, Kim K-S, Bergey EJ, Prasad PN (2002) Nanochemistry: synthesis and characterization of multifunctional nanoclinics for biological applications. Chem Mater 14:3715–3721
Li Y, Yan B, Deng CH, Yu WJ, Xu XQ, Yang PY (2007) Efficient on-chip proteolysis system based on functionalized magnetic silica microspheres. Proteomics 7:2330–2339
Liu CX, Liu Q, Guo CC (2010) Synthesis and Catalytic Abilities of Silica-coated Fe3O4 Nanoparticle Bonded Metalloporphyrins with Different Saturation Magnetization. Catal Lett 138:96–103
Lu X, Zhang HP, Leng Y, Fang L, Qu S, Feng B, Weng J, Huang N (2010) The effects of hydroxyl groups on Ca adsorption on rutile surfaces: a first-principles study. J Mater Sci Mater Med 21:1–10
Mittal A, Krishnan L, Gupta VK (2005) Use of waste materials—Bottom Ash and De-Oiled Soya, as potential adsorbents for the removal of Amaranth from aqueous solutions. J Hazard Mater 117:171–178
Murray CB, Norris DJ, Bawendi MG (1993) Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites. J Am Chem Soc 115:8706–8715
Namasivayam C, Yamuna RT (1999) Studies on chromium (III) removal from aqueous solution by adsorption onto biogas residual slurry and its application to tannery wastewater treatment. Water Air Soil Pollut 113:371–384
Namasivayam C, Jeyakumar R, Yamuna RT (1994) Dye removal from waste-water by adsorption on waste Fe(III)/Cr(III) hydroxide. Waste Manage 14:643–648
Namasivayam C, Yamuna RT, Jayanthi J (2003) Removal of methylene blue from wastewater by adsorption on cellulosic waste, orange peel. Cell Chem Technol 37:333–339
Ngah WSW, Hanafiah MAKM (2008) Removal of heavy metal ions from wastewater by chemically modified plant wastes as adsorbents: a review. Bioresource Technol 99:3935–3948
Peric J, Trgo M, Vukojevic-Medvidovic N (2004) Removal of zinc, copper and lead by natural zeolite – a comparison of adsorption isotherms. Water Res 38:1893–1899
Piaoping Y, Zewei Q, Zhiyao H, Chunxia L, Xiaojiao K, Ziyong C, Jun L (2009) A magnetic, luminescent and mesoporous core–shell structured composite material as drug carrier. Biomaterials 30:4786–4795
Reddad Z, Gerente C, Andres Y, Le-Cloirec P (2002) Adsorption of several metal ions onto a low-cost biosorbent: kinetic and equilibrium studies. Environ Sci Technol 36:2067–2073
Sharma P, Rajaram P, Tomar R (2008) Synthesis and morphological studies of nanocrystalline MOR type zeolite material. J Colloid Interface Sci 325:547–557
Shriver DF, Atkins PW, Langford CH (1990) Inorganic Chemistry, 1st edn. Freeman, New York
Srivastava NK, Majumder CB (2008) Novel biofiltration methods for the treatment of heavy metals from industrial wastewater. J Hazard Mater 151:1–8
Srivastava SK, Gupta VK, Dwivedi MK, Jain S (1995) Caesium PVC–crown (dibenzo-24-crown-8) based membrane sensor. Anal Proc 32:21–23
Taty-Costodes VC, Fauduet H, Porte C, Delacroix A (2003) Removal of Cd(II) and Pb(II) ions, from aqueous solutions, by adsorption onto sawdust of Pinus sylvestris. J Hazard Mater 105:121–142
Yang PP, Quan ZW, Lu LL, Huang SS, Lin J (2008) Bioactive, luminescent andmesoporous europium-doped hydroxyapatite as a drug carrier. Biomaterials 29:4341–4347
Zhao WR, Gu JL, Zhang LX, Chen HR, Shi JL (2005) Fabrication of uniform magnetic nanocomposite spheres with a magnetic core/mesoporous silica shell structure. J Am Chem Soc 127:8916–8917
Zhiya M, Yueping G, Huizhou L (2006) Superparamagnetic silica nanoparticles with immobilized metal affinity ligands for protein adsorption. J Magn Magn Mater 301:469–477
Acknowledgment
We gratefully thank the University Language Center, especially Mr. Fardin, for his cooperation in reviewing and editing this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Vinod Kumar Gupta
Rights and permissions
About this article
Cite this article
Padervand, M., Gholami, M.R. Removal of toxic heavy metal ions from waste water by functionalized magnetic core–zeolitic shell nanocomposites as adsorbents. Environ Sci Pollut Res 20, 3900–3909 (2013). https://doi.org/10.1007/s11356-012-1333-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-012-1333-y