
Abstract
Introduction
Magnetic Fe3O4 nanoparticles were prepared by coprecipitation and then were coated with SiO2 on the surface.
Materials and methods
Fe3O4@SiO2 composite microspheres were modified by KH570. Using molecular imprinting technology, atrazine magnetic molecularly imprinted polymer was prepared by using atrazine as template molecule, methacrylic acid as functional monomer and ethylene glycol dimethacrylate as cross-linkers. The morphology, composition and magnetic properties of magnetic nanoparticles were characterized. The recognition selectivity of polymer was studied for template molecule and simulation by UV spectrophotometry. The adsorption properties and selectivity ability were analyzed by Scatchard analysis.
Results
Scatchard linear regression analysis indicated that there are two binding sites of the target molecules. The magnetic molecularly imprinted polymer has been applied to the analysis of atrazine in real samples.
Conclusion
The results show that: the recovery rates and the relative standard deviation were 94.0∼98.7% and 2.1∼4.0% in corn, the recovery rates and the relative standard deviation were 88.7∼93.5% and 2.8∼7.2% in water.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Atrazine is one of the most widely used herbicides in cereal plants, such as corn, sorghum, and sugar-cane. As a consequence of its solubility, high chemical and biological stability in soils and aquifers, widespread use and considerable leaching potential, atrazine has been affected to environment (Zaya et al. 2011; Jablonowski et al. 2011). Typically, atrazine analysis is determined by gas chromatography (GC; Xu et al. 2007; Djozan and Ebrahimi 2008), high-performance liquid chromatography (HPLC) (Gamble et al. 2010), chromatography–mass spectrometry (Xu et al. 2007; Hu et al. 2011), and enzyme-linked immunosorbent assay (Lima et al. 2011) at present. The usually used pre-treatment technologies are liquid–liquid extraction and solid phase extraction in these methods. But these pre-treatment technologies have complex operations, large reagent consumption and low selectivity. Therefore, it is especially important to looking for separation and enrichment methods with high selectivity ability, low interference.
Molecularly imprinted polymers (MIPs) are obtained by polymerizing functional and cross-linking monomers around a template molecule, leading to a highly three-dimensional cross-linked network polymer. Polymerization by covalent, non-covalent or semi-covalent approaches has taken place. Then template molecule is remove and binding sites with shape, size and functionalities complementary to the target analyte are established. The MIPs are stable, robust and resistant to a wide range of pH, solvents, thermal stability, used long time (Chen et al. 2011), good specific adsorption properties and selectivity ability. It was used for selective enrichment and separate sample pre-treatment. But the process needs large consumption of solvents.
Magnetic nanoparticles have many superior characteristics such as small size, high coercivity, magnetic susceptibility and functional modification. They have some applications in a variety of fields such as catalysis, bioseparation, sensors, bioengineering, magnetic separation (Abareshi et al. 2010; Jun et al. 2008; Singh et al. 2011). However, magnetic nanoparticles are facile agglomerates, easily oxidized in air and unstable in aqueous solution during the synthesis and applications (Lu et al. 2007). For many applications, it is thus crucial to develop protection strategies to chemically stabilize the naked magnetic nanoparticles. These strategies comprise grafting of or coating with organic species, coating with an inorganic layer, such as silica or carbon. Coating silica is often applied to magnetic Fe3O4 nanoparticles to improve their biocompatibility, stability in aqueous conditions and functionality (Teleki et al. 2009; Huang et al. 2010). Furthermore, silica-coated or embedded magnetic Fe3O4 nanoparticles exhibit enhanced thermal stability.
Therefore, magnetic molecularly imprinted polymers (M-MIPs) are obtained by Fe3O4@SiO2 coated with molecular imprinting technique. It is noteworthy that the M-MIPs have not only excellent superparamagnetism of the magnetic nanoparticles, but also have high selectively recognized the analytes in the complex matrix. The M-MIP will be a convenient and quickly recognizable separation method with external magnetic field. Nowadays, the M-MIP synthesized by using the Fe3O4@SiO2 coated has been reported in the some literatures. The M-MIPs particles for estrone using template, dimethyl sulfoxide as porogen and with Fe3O4@SiO2 particles was reported by Wang et al. (2009). Li et al. (2010) reported that magnetic molecularly imprinted polymers films with RAFT polymerization on Fe3O4@SiO2 surface. The core-shell magnetic beads showed a high affinity, selectivity, and easy separation behavior for BPA. Jing et al. (2010) reported the M-MIP films are obtained using lysozyme as a template, methacrylic acid and acrylamide as functional co-monomers, and magnetic Fe3O4@SiO2 particles.
In this work, the superparamagnetic core-shell beads were attempted to synthesize for the recognition of atrazine. The Fe3O4 nanoparticles were synthesized by the coprecipitation method and then covered with SiO2 by sol–gel method. The preparation method of M-MIP beads is schematically illustrated in Scheme 1. The M-MIPs were attracted by a magnet on the bottom of reaction bottle when extraction and separation. The M-MIP beads were applied to corn and river water samples for atrazine, and to solve the shortcomings of traditional solid phase extraction, such as the column needs to be filled, low selectivity and large consumption of solvents.

Preparation process of Fe3O4@SiO2 molecularly imprinted polymer
2 Experiment section
2.1 Materials
Ferrous sulfate heptahydrate (FeSO4•7H2O), ammonium hydroxide, and tetraethyl orthosilicate (TEOS) were purchased from Jiaxing Jing Bol Reagent Company (Jiaxing, China); ferric chloride hexahydrate (FeCl3•6H2O), methacrylic acid (MAA) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China); toluene and methanol were purchased from Hangzhou Chemical Reagent Co., Ltd (Hangzhou, China); 3-methacryloxypropyltrimethoxysilane (MPS, KH570), ethylene dimethacrylate (EGDMA), 2,2′-azobis(2-methylpropionitrile) (AIBN), cyanazine, melamine were purchased from Shanghai Pure Industrial Co., Ltd (Shanghai, China); atrazine was purchased from Jintan Weidelong Chemical Co., Ltd (Jintan, China); dimethyl sulfoxide (DMSO) was obtained from Hangzhou Shuanglin Chemical Reagent factory (Hangzhou, China); citric acid was obtained from Shanghai Chemical Reagent Co., Ltd (Shanghai, China); ethanol was obtained from Lin'an Qingshan Chemical Reagent Factory (Linan, China); hydrochloric acid was obtained from Zhejiang Juhua Xinlian Chemical Co., Ltd (Quzhou, China); acetic acid was obtained from Zhejiang Zhongxing Chemical Reagent Co., Ltd (Jinhua, China); ultrapure water was made by a laboratory (Human, Korea); corn was purchased from a local supermarket; and environmental water was obtained from Jiaxing river.
2.2 Synthesis of the M-MIPs
To synthesize Iron oxide nanoparticles, FeSO4•7H2O and FeCl3•6H2O were prepared by dissolving in ultrapure water (100 ml) with under nitrogen for 30 min to remove oxygen. The molar ratio of Fe2+ and Fe3+ was 1:1.75. Then, 10 ml of ammonia aqueous solution was dropwise added into flask with vigorous stirring and continue to reaction in oil bath at 60°C. After 2 h, 1.0 g citric acid was added into the solution and reaction was kept for 1.5 h. The black sediment was collected by applying a magnet and rinsed several times with ethanol and highly purified water until it was neutral. Finally, magnetic nanoparticles obtained were dried under vacuum at 60°C for overnight.
Fe3O4@SiO2 microspheres were prepared according to the sol–gel method. Typically, Fe3O4 (0.1 g) was dispersed in ethanol (60 ml) and ultrapure water(10 ml) by sonication for 15 min, followed by the addition of ammonium hydroxide (1 ml) and TEOS (2 ml) sequentially. The mixture solution reacted for 24 h at the room temperature under a continuous stirring. The synthesized product was separated by a magnet and washed with ethanol and deionized water several times, and dried under vacuum at 60°C. Subsequently, Fe3O4@SiO2 microspheres were added into hydrochloric acid solution (10%), the mixture was heated at reflux for 6 h. The product was washed several times with ethanol and highly purified water, and dried in vacuum at 60°C.
To prepare the Fe3O4@SiO2 particles modified with MPS, 0.3 g Fe3O4@SiO2 activation microspheres were dispersed in 100 ml of anhydrous toluene, followed by the addition of 2 ml of MPS. Then, the mixture was incubated at 60°C for 5 h under nitrogen protection. The Fe3O4@SiO2 microspheres modified by MPS (Fe3O4@SiO2-MPS) was obtained by magnetic separation and rinsed with DMSO and ethanol several times, respectively.
Then M-MIP films were synthesized as follows process. The template (atrazine, 1 mmol) and functional monomer (MAA, 4 mmol) were dispersed in toluene (15 ml) by sonication, and the mixture has been shaken for overnight. Then, the Fe3O4@SiO2-MPS particles (0.2 g), the cross-linking monomer (EGDMA, 8 mmol), and the initiator (AIBN, 0.09 g) were added to the mixture, respectively. Subsequently, the solution was deoxygenated by purging with nitrogen for 30 min, sealed and shaken at 60°C for 24 h. Upon completion, template molecule of the resulting product were removed with a mixture solvent of methanol and acetic acid (9:1, v/v) by Soxhlet extraction until the eluent was free from atrazine through detected by UV/Vis spectrophotometer. Finally, the M-MIPs obtained were washed with methanol to remove the remaining acetic acid and dried in the vacuum. As a reference, magnetic non-imprinted polymer (M-NIP) were also prepared using the same procedures but without adding atrazine templates.
2.3 Atrazine adsorption experiments
To investigate the adsorption capacity, M-MIP was added to 10 ml methanol solution with various concentrations of atrazine. The mixture was shaken continuously at room temperature for a long time to facilitate the adsorption of atrazine onto the M-MIP sorbent. Then, the M-MIP beads were separated by a magnet. The supernatant solution was measured using the UV-spectrometer. The adsorption capacity (Q) was calculated from Eq. 1 with the experimental data. The same procedure was performed for the M-NIP.
2.4 Selectivity experiments
Competitive recognition studies were performed with atrazine and the structurally similar compound cyanazine and melamine. The M-MIP (10 mg) films were placed in 10 ml of methanol solutions containing 80 μmol/L of atrazine, cyanazine or melamine. The mixture was rocked for 5 h at room temperature, and the polymers were isolated using a magnet. The concentrations of atrazine, cyanazine or melamine were detected by UV/Vis spectrophotometer and the distribution coefficient and selectivity coefficient were calculated. The M-NIP was also examined with the same procedure.
2.5 Instrumentation and characterizations
Fourier transform infrared (FT-IR) spectra of the nanoparticles were recorded with NEXUS-470 (Nicolet, USA) spectrometer using KBr method. Thermogravimetric analysis (TGA) was performed by a STA-409PC instrument (Germany) from room temperature to 800°C with a heating rate of 20°C min−1 in a nitrogen flow. Magnetic properties were measured with a LakeShore 7407 (Lakeshore, USA) vibrating sample magnetometry (VSM) at room temperature. The structure of the powder samples was characterized by a DX-2600 X-ray diffractometer (XRD; Dandong, China). The products sizes were characterized by a JEM-200CX transmission electron microscope (TEM; JEOL, Japan). And the samples for TEM measurements were prepared by placing one drop of the samples on copper grids coated with carbon. The concentrations of all solutions were detected on a UV-2550 UV/Vis spectrometer (Shimadzu, Japan).
The low concentrations of atrazine solutions were assayed on an Agilent-1200 HPLC (Agilent, USA). The HPLC system equipped with a G1314B UV/Vis detector and G1328B manual-sampler. The stationary phase consisted of a column (5 μm, 4.6 mm × 250 mm) packed with extend-C18. The detection wavelength was 270 nm for atrazine. The mobile phase consisted of water–methanol (70:30, v/v) for corn and water–methanol (65:35, v/v) for water. The column was eluted at a flow rate of 1.0 ml min−1 at 35°C.
3 Results and discussion
3.1 Characterization of magnetic microspheres
3.1.1 FT-IR spectra
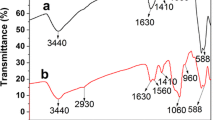
The FT-IR spectra of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-MPS, and M-MIP were given in Fig. 1. The characteristic absorption band of Fe–O situated at 580 cm−1 in each curves. The strong peak at 1,096 cm−1 was attributed to the asymmetry stretching vibration of Si–O–Si bonds. Other absorption bands, such as the Si–O bending vibration peak at 799 cm−1 and the Si–O–H vibration peak at around 954 cm−1 were appeared in the Fe3O4@SiO2 sample (Fig. 1b). Such results confirmed that SiO2 was indeed coated onto the surface of Fe3O4 nanoparticles. The typical peak of 1,730 cm−1 represents the stretching vibration of carbonyl groups of the MPS in Fig. 1c. Upon comparing M-MIP to Fe3O4@SiO2-MPS, a bond absorption band at 2,970 cm−1 of the M-MIP corresponded to the C–H aromatic stretching vibration of EGDMA. Then, the peak of C–H at 1,390 cm−1 symmetric bending vibration from methyl groups and C═O bond at 1,730 cm−1 stretching vibration increase observably. The appearances of these bands were confirmed that the polymerization was successful.

FT-IR spectra of Fe3O4(a), Fe3O4@SiO2(b), Fe3O4@SiO2-MPS(c), and M-MIP(d)
3.1.2 Thermal stability analysis
Figure 2 depicted the TGA curves of the Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2-MPS (c), and M-MIP (d). As shown in Fig. 2a illustrated that the Fe3O4 (a), Fe3O4@SiO2 (b), and Fe3O4@SiO2-MPS (c) microspheres have good thermal stability. From room temperature to 800°C, there is only little lost for about 8%. In details, the rate of weight loss for Fe3O4 (a), Fe3O4@SiO2 (b) and Fe3O4@SiO2-MPS (c) owed to remnant ethanol the dehydration in the layer of samples under below 150°C. When the temperature was changed from 400°C to 600°C, the decreased weight might be due to decomposition of organic matter in Fe3O4@SiO2 (b) and Fe3O4@SiO2-MPS (c). Especially the weight loss rate of Fe3O4@SiO2-MPS (c) was much higher than the Fe3O4@SiO2 (b) at nearby 500°C, which demonstrated existence of MPS.

TGA curves of the Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2-MPS (c) and M-MIP, (d) magnetic microspheres
From Fig. 2b, it is obvious that M-MIP had a rapid weight loss rate from 300°C to 500°C, and the decreased weight of M-MIP was approximately 84.4%. The weight loss should be the decomposition of MIP.
3.1.3 Magnetic properties
The magnetic properties of the Fe3O4 nanoparticles (a), Fe3O4@SiO2 (b), and Fe3O4@SiO2-MPS (c) were characterized by VSM at room temperature, and the results are shown in Fig. 3a. The values of saturation magnetization (Ms), coercivity (Hc), and squareness (Sr = remnant magnetization (Mr)/Ms) for the Fe3O4 were 63.76 emu g−1, 32.40 Oe, and 0.035, respectively. The saturation magnetization value of Fe3O4 was similar to that from the literature (Ji et al. 2009; Kan et al. 2009). And these values were smaller than that of the theoretical specific saturation magnetization of bulk magnetite (92 emu/g; Zaitsev et al. 1999). The value of saturation magnetization was decreased due to the small particle surface effect. The saturation magnetization value of the Fe3O4@SiO2 was far less than that of the pure Fe3O4 nanoparticles. The decrease in magnetization value is most likely attributed to the existence of silica shells on the surface of Fe3O4 nanoparticles, leading to a weakening of the magnetic moment. The Fe3O4@SiO2 exhibited slightly enhanced magnetic properties after modified by MPS.

Magnetic hysteresis loop and photographs of the separation process of magnetic microspheres
The magnetic hysteresis loop of the M-MIP is illustrated in Fig. 3b. The values of Ms, remnant magnetization (Mr) and Hc for the M-MIP were 1.15 emu g−1, 25.24 emu g−1 and 41.51 Oe, respectively. The remnant magnetization value of the M-MIP is smaller compared with the pure Fe3O4 nanoparticles. So the agminate M-MIP can renewably disperse rapidly after the magnetic field is removed. Figure 3B (inset) shows the photographs of the separation process of M-MIP beads. In the absence of an external magnetic field, a yellow homogeneous dispersion exists. When an external magnetic field was applied, the black particles were attracted to the wall of vial and the dispersion became clear and transparent in a short time. As a result, the M-MIP particles could be easily and quickly separated from a suspension.
3.1.4 X-ray diffraction patterns
The structural properties of Fe3O4 nanoparticles, Fe3O4@SiO2, Fe3O4@SiO2-MPS and M-MIP were analysized by XRD. As shown in Fig. 4, the discernible six characteristic peaks for Fe3O4 (2θ = 30.2°, 35.6°, 43.3°, 53.5°, 57.2°, and 62.8°) were observed for all the samples, and the peak positions could be indexed to (220), (311), (400), (422), (511) and (440), respectively, which match well with the database of magnetite in the JCPDS-International Center (JCPDS Card: 19-0629). The peak positions were unchanged during the synthesized process, which indicating that the crystalline structure of the magnetite was essentially maintained. The diffraction peak with 2θ = 22° can be assigned to the amorphous SiO2 for Fe3O4@SiO2, Fe3O4@SiO2-MPS and M-MIP (Fig. 4b, c and d). It is also showed that the Fe3O4 nanoparticles were indeed incorporated into all of the samples. However, it is insufficient to exclude the presence of maghemite (γ-Fe2O3), and there are probably two types of iron oxide particles in this dispersion: maghemite and magnetite. XRD analysis cannot discriminate between magnetite and its maghemite because both diffraction patterns overlap and are nearly identical (Li et al. 2009). The trace amounts of maghemite could be attributed to the oxidation of Fe3O4 to γ-Fe2O3 during the coprecipitation and modification process.

XRD patterns of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2-MPS(c), and M-MIP (d)
3.1.5 Morphological characteristics of the synthesized magnetic nanoparticles
The morphological structure of Fe3O4 nanoparticles, Fe3O4@SiO2, Fe3O4@SiO2-MPS and M-MIP were provided by TEM as shown in Fig. 5. The image of the uncoated Fe3O4 microsphere reveals a homogeneous size distribution with a mean diameter of about 15 nm by TEM in Fig. 5a. Figure 5b shows the TEM image of the Fe3O4@SiO2 with average diameter of 200 nm. It is obvious seen that the black Fe3O4 nanoparticles existed in the internal of the silica layer, which revealed that the magnetite nanoparticles were fully coated by the silica. The morphological features of the Fe3O4@SiO2 nanoparticles (Fig. 5b) were similar to that of the Fe3O4@SiO2-MPS (Fig. 5c). Figure 5d clearly shows the thickness of shell membrane of core-shell structure increases slightly, which can be attributed to the fact that the reaction combined of organic matters on the particles surface. These observations also confirmed that the M-MIPs beads had been successfully prepared.

TEM micrograph of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2-MPS (c), and M-MIPs (d)
3.2 Adsorption properties of M-MIPs
3.2.1 The optimization of the magnetic polymer dosage
Figure 6 shows the adsorption capacity of the polymer rapidly increased with increase of the dosage in the less than 10 mg. After more than 10 mg, the adsorption capacity of MIPs started to decreased. With the increase of M-MIP, we found the adsorption capacity was decreased. We thought that the M-MIP simultaneously had an obvious aggregation between the nanoparticles when the dosage was more than 10 mg. So some binding active sites were embedded. The adsorption capacity will drop with the absorbent increased by using Eq. 1. Hence, 10 mg was chosen for optimum dosage.

The dosage optimization of molecularly imprinted polymer
3.2.2 The best adsorption time of the magnetic polymer and adsorption kinetics
As can be seen from Fig. 7, the adsorption capacity of the M-MIPs and M-NIPs increased with the increasing of adsorption time, and reached the equilibrium adsorption for 5 h. Simultaneously, The M-MIPs had much higher recognition ability than the M-NIPs at all rebinding processes.

The adsorption capacity-adsorption time curve of M-MIP and M-NIP microspheres
The kinetics of adsorption is important because it controls the process efficiency. For investigating the adsorption kinetics of atrazine onto M-MIPs, the pseudo-first-order and pseudo-second-order kinetic models were applied to the experimental data. The adsorption process of the M-MIPs was confirmed by according to the value of correlation coefficients.
The kinetic data were calculated by using Eqs. 2 and 3. The rate constants of adsorption along with linear regression correlation coefficients were presented in Table 1. The value of correlation coefficient for pseudo-second-order kinetic model is better than that of the pseudo-first-order. From Table 1, it is evident that chemical process could be the rate-limiting step in the adsorption process for atrazine.
In the equations above, Q e (in milligrams per gram) represents the adsorbed amount of atrazine at equilibrium and Q t (in milligrams per gram) at time t (h). Q 1 (in milligrams per gram) and Q 2 (in milligrams per gram) are the pseudo-first-order and pseudo-second-order theory adsorption amount. k 1 (in per hour) and k 2 (in grams per milligram per hour) are the rate constant of pseudo-first-order and pseudo-second-order, respectively.
3.2.3 The Scatchard analysis and adsorption isotherm
The adsorption capacity is an important factor to evaluate the special binding and selective recognition of molecular imprinting technique. The rebinding experiments were performed by varying the initial concentration of atrazine (from 10 to 100 μmol L−1) in the presence of 10 mg M-MIP or M-NIP at room temperature. As shown in Fig. 8, atrazine adsorbed on the M-MIP increased with increase of the initial atrazine concentration; the adsorption curve became increased slowly when C o exceeded 80 μmol L−1. Furthermore, the M-MIP exhibited significantly higher atrazine loading than the M-NIP. It is illustrated that the specific adsorption of sites are incorporated into M-MIP for the recognition of atrazine.

The relationship between the adsorption capacity and concentrations of M-MIP and M-NIP
In order to further study the binding properties of the M-MIP beads, Scatchard analysis was performed by using the saturation binding data. The values of maximum adsorption capacity and K d were obtained by using Eq. 4. It can be seen clearly in Fig. 9, the relationship between Q/C and Q could be expressed using two straight lines, which illustrated that two kinds of different binding sites existed in M-MIPs. The values of K d and Q max were determined from the slopes and intercepts of the linear regression formulas. The Q max and K d of the high affinity sites were 2.497 μmol g−1 and 46.54 μmol L−1, while Q max and K d of the low affinity sites were 1.341 and 10.06 μmol L−1, respectively.
Where K d is the dissociation constant and Q max is the apparent maximum number of binding sites. C e is the free atrazine concentration at equilibrium.

Scatchard analysis of M-MIP
The equilibrium data were further analyzed using the Langmuir and Freundlich isotherm models, respectively. The adsorption type for M-MIPs is determined by the saturated adsorption capacity and linearized coefficient.
The isotherm parameters obtained from linear analysis of the Eqs. 5 and 6 are given in Table 2. The results showed that the Freundlich isotherm model was more suitable for the experimental data than that by the Langmuir isotherm, for the high value of the correlation coefficients. It is implied that the adsorption takes place at specific homogeneous sites and further adsorption sites.
Here, C e (in micromoles per liter) and Q e (in micromoles per gram) are atrazine concentration and adsorption capacity at adsorption equilibrium. Q m (in micromoles per gram), the theoretical maximum adsorption capacity. K (in liters per micromole) is the Langmuir constant. K f and 1/n are the Freundlich constants, which are indicators of the adsorption capacity and adsorption intensity.
3.2.4 Selectivity adsorption of M-MIP
To further investigate the selective recognition properties of the M-MIP, the structurally similar compound cyanazine and melamine were selected to act as the competitors. The experiment was performed by adding M-MIP into the each solution. To test the selective recognition of MIP, the same experiments were carried out with M-NIP. The adsorption capacity (Q e), the distribution coefficient (K d), the imprinting factor (IF), the selectivity coefficient (k) (Han et al. 2005), and the relative selectivity coefficient (k′) (Han et al. 2005) were obtained from the selectivity adsorption experiments and showed in Table 3. The K d indicated the adsorption ability of a substance. K embodied selectivity when the sorbent is presented between target molecule and reference substances, and k′ reveals the selective by between MIPs and NIPs for one particular substance.
As can be seen from Table 3, the values of IF were calculated to be 1.7 of atrazine for M-MIP films, and the selectivity parameters of M-MIP were higher than that of the structural analogues. It can be implied that the M-MIP provided high selectivity to atrazine, which illuminated specific recognition sites and chemical structure for template molecule are created during the course of imprinting. Moreover, there was no obvious difference in absorbing cyanazine and melamine for M-MIP.
3.3 Analytical performances
A series of low concentration of the atrazine standard solution were prepared by using solution of corn extract, which analyzed by HPLC method with the optimized chromatographic conditions. And the standard curve could be expressed by the peak area (A) on the concentration (in micrograms per milliliter ). The results indicated that the linearity of the method was investigated at the range of 0.1–2.0 μg mL−1 with squared coefficients of correlation R = 0.9997, and the linear equation was A = 32.29235 × C − 0.53602. Additionally, the limit of detection (LOD; 3σ/K, n = 11) for atrazine was found to be 5 μg L−1. Comparable to the value of literature, the data of LOD in this work is even better or similar. It is reported that the limit of detection was 5 μg L−1 and 0.01 μg mL−1 by Zhang et al. (2009) and Koohpaei et al. (2009), respectively.
The atrazine standard solution (50 μg mL−1) was diluted with the solution of river water. The peak areas (A) with corresponding concentration (C) were obtained by the analysis of the samples which were determined by HPLC under the optimized chromatographic conditions. The results showed that, good linearity was achieved by the relationship between A and C in the range from 0.1 to 1.5 μg mL−1, and the linear equation was A = 27.85648 × C − 0.34355. Moreover, the correlation coefficients (R) and the limit of detection (LOD, 3σ/K, n = 11) were R = 0.9995 and 3 μg L−1, respectively. These data with the developed method in this wok showed comparable or even better results against reported previously method (Djozan and Ebrahimi 2008; Zhang et al. 2009; Koohpaei et al. 2009).
3.4 The application in real samples analysis
3.4.1 Analysis of corn sample
To further validate the feasibility of the method, corn sample was selected for spiked analyzed. Initially, the sample extraction was performed by spiking atrazine solution at three different concentrations of 0.1, 0.25 and 0.5 μg mL−1 after quiescence 12 h. These extraction solutions of corn were subjected to enrichment by the M-MIP and M-NIP beads, and were eluted by elution solution of methanol and then analyzed by HPLC. The recovery of atrazine and the relative standard deviation can be observed in Table 4. It could be seen that the recovery of atrazine by the M-MIP is high than that of the M-NIP, which was identical with the results of the previous adsorption results. The results indicated that the M-MIP can be applied in determination of trace analytes from complicated samples.
Under the optimized experimental conditions, the molecular recognition effect were determined for corn sample with 0.25 μg mL−1 atrazine after extraction with M-MIP and M-NIP, and obtained for corn sample without spiked atrazine after extraction with M-MIP, respectively. The chromatograms of the real and spiked corn samples are shown in Fig. 10. As shown in Fig. 10, atrazine could not be detected in the real sample. In addition, the utilization of the M-MIP resulted in much better extraction than that of the M-NIP, which illustrated the high selectivity and recognition was observed for the MIP extraction.

The chromatogram of the corn sample after extraction: the spiked corn sample with 0.25 μg mL−1 atrazine after extraction with M-MIP (a), M-NIP (b), without spiked atrazine after extraction with M-MIP (c)
3.4.2 Environmental water sample analysis
It was further applied to analyze atrazine in river water for demonstrate the applicability of the method. The river water was added by spiking atrazine solution at three levels (0.1, 0.25, and 0.5 μg mL−1). These mixture solutions were subjected to enrichment by the M-MIP and M-NIP beads, and were eluted by elution solution of methanol under the experimental conditions. Then, the recovery of atrazine, the standard deviation (SD), and the relative standard deviation (RSD) were calculated by analyzing the elution solution using HPLC, and shown in Table 5. The results showed that the spiked recoveries of atrazine by using M-MIP were changed from 88.7% to 93.5%, and the RSD (N = 3) were in the range of 2.8–7.2%. Simultaneously, the recovery of atrazine by the M-MIP is high than that of the M-NIP, the resulted demonstrated that the good selection of adsorption of the M-MIP for atrazine in the water system.
4 Conclusions
In the study, the M-MIPs using atrazine as a template were prepared based on surface modification of the Fe3O4@SiO2 nanoparticles by using molecularly imprinting technique. The obtained magnetic microspheres were characterized by FT-IR, TGA, VSM, XRD, and TEM. In the rebinding experiment, high adsorption capacity and good selectivity for atrazine were demonstrated by the M-MIP beads. Furthermore, the M-MIPs were successfully applied to selective enrichment coupled with HPLC analysis for triazines in spiked samples. In conclusion, the M-MIPs not only provided a convenient separation with the magnetic nanoparticles, but it also overcame problems of low selectivity with MIP. So a novel method combining M-MIP with HPLC analysis was established to determine the atrazine.
References
Abareshi M, Goharshadi EK, Zebarjad SM, Fadafan HK, Youssefi A (2010) Fabrication, characterization and measurement of thermal conductivity of Fe3O4 nanofluids. J Magn Magn Mater 322:3895–3901
Chen LX, Xu SF, Li JH (2011) Recent advances in molecular imprinting technology: current status, challenges and highlighted applications. Chem Soc Rev 40:2922–2942
Djozan D, Ebrahimi B (2008) Preparation of new solid phase micro extraction fiber on the basis of atrazine-molecular imprinted polymer: application for GC and GC/MS screening of triazine herbicides in water, rice and onion. Anal Chim Acta 616:152–159
Gamble DS, Webster GB, Lamoureux M (2010) Quantitative prediction of atrazine sorption in a Manitoba soil using conventional chemical kinetics instead of empirical parameters. J Phys Chem C 114:20055–20061
Han DM, Fang GZ, Yan XP (2005) Preparation and evaluation of a molecularly imprinted sol–gel material for on-line solid-phase extraction coupled with high performance liquid chromatography for the determination of trace pentachlorophenol in water samples. J Chromatogr A 1100:131–136
Hu XH, Wang XW, Zhou Q, Wu YM, Xu QX (2011) Research of qualitative ELISA and GC-MS for atrazine in soil. Biotechnol Mater Eng 183:990–993
Huang J, Zhao R, Wang H, Zhao WQ, Ding LY (2010) Immobilization of glucose oxidase on Fe3O4/SiO2 magnetic nanoparticles. Biotechnol Lett 32:817–821
Jablonowski ND, Schäffer A, Burauel P (2011) Still present after all these years: persistence plus potential toxicity raise questions about the use of atrazine. Environ Sci Pollut Res 18:328–331
Ji YS, Yin JJ, Xu ZG, Zhao CD, Huang HY, Zhang HX, Wang CM (2009) Preparation of magnetic molecularly imprinted polymer for rapid determination of bisphenol A in environmental water and milk samples. Anal Bioanal Chem 395:1125–1133
Jing T, Du HR, Dai Q, Xia H, Niu JW, Hao QL, Mei SR, Zhou YK (2010) Magnetic molecularly imprinted nanoparticles for recognition of lysozyme. Biosens Bioelectron 26:301–306
Jun YW, Seo JW, Cheon JW (2008) Nanoscaling laws of magnetic manoparticles and their applicabilities in biomedical sciences. Acc Chem Res 41:179–189
Kan XW, Geng ZR, Zhao Y, Wang ZL, Zhu JJ (2009) Magnetic molecularly imprinted polymer for aspirin recognition and controlled release. Nanotechnology 20:1–7
Koohpaei A-R, Shahtaheri S-J, Ganjali M-R, Forushani A-R, Golbabaei F (2009) Optimization of solid-phase extraction using developed modern sorbent for trace determination of ametryn in environmental matrices. J Hazard Mater 170:1247–1255
Li L, He XW, Chen LX, Zhang YK (2009) Preparation of core-shell magnetic molecularly imprinted polymer nanoparticles for recognition of bovine hemoglobin. Chem-Asian J 4:286–293
Li Y, Li X, Chu J, Dong CK, Qi JY, Yuan YX (2010) Synthesis of core-shell magnetic molecular imprinted polymer by the surface RAFT polymerization for the fast and selective removal of endocrine disrupting chemicals from aqueous solutions. Environ Pollut 158:2317–2323
Lima DL, Silva CP, Schneider RJ, Esteves VI (2011) Development of an ELISA procedure to study sorption of atrazine onto a sewage sludge-amended luvisol soil. Talanta 85:1494–1499
Lu A-H, Salabas E-L, Schüth F (2007) Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew Chem Int Ed 46:1222–1244
Singh KP, Singh AK, Singh UV, Verma P (2011) Optimizing removal of ibuprofen from water by magnetic nanocomposite using Box–Behnken design. Environ Sci Pollut Res
Teleki A, Suter M, Kidambi PR, Ergeneman O, Krumeich F, Nelson BJ, Pratsinis SE (2009) Hermetically coated superparamagnetic Fe2O3 particles with SiO2 nanofilms. Chem Mater 21:2094–2100
Wang X, Wang LY, He XW, Zhang YK, Chen LX (2009) A molecularly imprinted polymer-coated nanocomposite of magnetic nanoparticles for estrone recognition. Talanta 78:327–332
Xu RJ, Wei FS, Wang YG, Hu W, Ye XQ, Xu GW (2007) Determination of atrazine and its metabolites in human urines using gas chromatography. Chinese J Chromatogr/Zhongguo Hua Xue Hui 25:758–761
Zaitsev V-S, Filimonov D-S, Presnyakov I-A, Gambino R-J, Chu B (1999) Physical and chemical properties of magnetite and magnetite-polymer nanoparticles and their colloidal dispersions. J Colloid Interf Sci 212:49–57
Zaya RM, Amini Z, Whitaker AS, Kohler SL, Ide CF (2011) Atrazine exposure affects growth, body condition and liver health in Xenopus laevis tadpoles. Aquat Toxicol 104:243–253
Zhang Y, Liu RJ, Hu YL, Li GK (2009) Microwave heating in preparation of magnetic molecularly imprinted polymer beads for trace triazines analysis in complicated samples. Anal Chem 81:967–976
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 21177049, No. 51103063), the Program for Science and Technology of Zhejiang (No. 2011C22096, No. 2011C37033), the Program for Science and Technology Innovation of Zhejiang for undergraduate (No. 2011R417020) and the Program for Science and Technology of Jiaxing (Nos. 2010AY1081, 2011AY1028, 2011AY1007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article
Cite this article
Men, HF., Liu, HQ., Zhang, ZL. et al. Synthesis, properties and application research of atrazine Fe3O4@SiO2 magnetic molecularly imprinted polymer. Environ Sci Pollut Res 19, 2271–2280 (2012). https://doi.org/10.1007/s11356-011-0732-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-011-0732-9