
Abstract
Background, aim and scope
Photocatalytic oxidation using UV irradiation of TiO2 has been studied extensively and has many potential industrial applications, including the degradation of recalcitrant contaminants in water and wastewater treatment. A limiting factor in the oxidation process is the recombination of conduction band electrons (e− cb) with electron holes (h +vb ) on the irradiated TiO2 surface; thus, in aqueous conditions, the presence of an effective electron scavenger will be beneficial to the efficiency of the oxidation process. Ferrate (FeO 2−4 ) has received much recent attention as a water treatment chemical since it behaves simultaneously as an oxidant and coagulant. The combination of ferrate [Fe(VI)] with UV/TiO2 photocatalysis offers an oxidation synergism arising from the Fe(VI) scavenging of e− cb and the corresponding beneficial formation of Fe(V) from the Fe(VI) reduction. This paper reviews recent studies concerning the photocatalytic oxidation of problematic pollutants with and without ferrate.
Materials and methods
The paper reviews the published results of laboratory experiments designed to follow the photocatalytic degradation of selected contaminants of environmental significance and the influence of the experimental conditions (e.g. pH, reactant concentrations and dissolved oxygen). The specific compounds are as follows: ammonia, cyanate, formic acid, bisphenol-A, dibutyl- and dimethyl-phthalate and microcystin-LR. The principal focus in these studies has been on the rates of reaction rather than on reaction pathways and products.
Results
The presence of UV/TiO2 accelerates the chemical reduction of ferrate, and the reduction rate decreases with pH owing to deprotonation of ferrate ion. For all the selected contaminant substances, the photocatalytic oxidation rate was greater in the presence of ferrate, and this was believed to be synergistic rather than additive. The presence of dissolved oxygen in solution reduced the degradation rate of dimethyl phthalate in the ferrate/photocatalysis system. In the study of microcystin-LR, it was evident that an optimal ferrate concentration exists, whereby higher Fe(VI) concentrations above the optimum leads to a reduction in microcystin-LR degradation. In addition, the rate of microcystin-LR degradation was found to be strongly dependent on pH and was greatest at pH 6.
Discussion
The initial rate of photocatalytic reduction under different conditions was analysed using a Langmuirian form. Decrease in rates in the presence of dissolved oxygen may be due to competition between oxygen and ferrate as electron scavengers and to non-productive radical species interactions. The reaction between ferrate(VI) and microcystins-LR in the pH range of 6.0–10.0 is most likely controlled by the protonated Fe(VI) species, HFeO −4 .
Conclusions
The photocatalytic oxidation of selected, recalcitrant contaminants was found to be significantly greater in the presence of ferrate, arising from the role of ferrate in inhibiting the h +vb –e− cb pair recombination on TiO2 surfaces and the corresponding generation of highly oxidative Fe(V) species. The performance of the ferrate/photocatalysis system is strongly influenced by the reaction conditions, particularly the pH and dissolved oxygen concentration, arising from the complex nature of the interactions between the catalyst and the solution. Overall, the treatment performance of the Fe(VI)–TiO2–UV system is generally superior to alternative chemical oxidation methods.
Recommendations and perspectives
The formation of intermediate Fe(V) species in the photocatalytic reduction of ferrate(VI) requires confirmation, and a method involving electron paramagnetic resonance spectroscopy could be applied for this. The reactivity of Fe(V) with the selected contaminants is required in order to better understand the role of ferrate in the Fe(VI)–TiO2–UV oxidation system. To increase the practical utility of the system, it is recommended that future studies involving the photocatalytic oxidation of pollutants in the presence of ferrate(VI) should focus on developing modified TiO2 surfaces that are photocatalytic under visible light conditions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Background, aim and scope
Titanium dioxide (TiO2) is considered to be one of the most efficient and environmentally benign photocatalysts, and it has been widely used in paints, toothpaste, ointments, sunscreens and as a pigment (Chen and Mao 2007). The phenomenon of photocatalytic splitting of water on a TiO2 electrode under UV light was first reported in the early 1970s, and since then, several papers have appeared in the literature describing many promising applications in areas such as photovoltaics, sensors and in the photodegradation of pollutants (Fujishima and Honda 1972; Hoffmann et al. 1995; Grätzel 2001; Chae et al. 2003; Cozzoli et al. 2003; Dutta et al. 2005; Liu et al. 2006; Seluck and Bekbolet 2008). A summary of the reactions upon illumination of TiO2 under dissolved oxygen conditions is given by equations T1–T8 in Table 1. These reactions generate hydroxyl (•OH) and superoxide (O2 -•) radicals which are the primary reactive species in the photocatalytic oxidation of pollutants. However, it was found that the electron holes at the catalyst surface (Eq. T1 in Table 1) preferentially recombine with electrons in surface sites of mixed-phase Degussa P25 TiO2 (Eq. T2 in Table 1; Hurum et al. 2005). This results in a reduction in the efficiency of photocatalytic processes. Hence, reactions that either consume \( {\text{h}}^{+}_{\text{vb}} \,{\text{or}}\,{\text{e}}^{ - }_{\text{cb}} \) can enhance the photocatalytic activity of TiO2. Molecular O2, silver(I), mercury(II) and chromium(VI) have been used in combination with photocatalytic processes (Prairie et al. 1993; Linesebigler et al. 1995). Iron in its +6 oxidation state, ferrate(VI) (Fe(VI), FeVIO 2−4 ) can serve as an alternative to undesirable (toxic) metal ions to increase the photocatalytic efficiency.
Fe(VI) has been of considerable research interest because of its role as an environmentally friendly oxidant and disinfectant in remediation processes (Sharma 2002a, 2004, 2007; Jiang and Lloyd 2002; Yuan et al. 2002; Sharma et al. 2006, 2008; Jiang 2007; Yngard et al. 2008). Fe(VI) species are strong oxidising agents which can be seen from the reduction potential of reactions 1 and 2 as follows, in acidic and alkaline solutions, respectively.
The reduction potential of ferrate(VI) is more positive than the TiO2 conduction band electron’s potential (E cb = −0.6 to −0.8 V) in basic solution (Chenthamarakshan et al. 2000). It is likely that the heterogeneous photocatalytic reduction of Fe(VI) takes place through three one-electron steps that would result in the sequential formation of iron in +5 and +4 oxidation states [ferrate(V) and ferrate(IV)] (Eqs. F1–F3 in Table 1). Both of these oxidation states of iron are much more reactive than ferrate(VI) (Sharma 2002a, 2004, 2008; Cabelli and Sharma 2008; Sharma et al. 2001a, 2002, 2005). The comparison of reactivity of ferrate(VI) and ferrate(V) is given in Table 2. Ferrate(V) reacts orders of magnitude faster with inorganic and organic molecules than ferrate(VI) does. Ferrate(V) and ferrate(IV) thus have the ability to oxidise pollutants that cannot be easily oxidised by ferrate(VI). This paper reviews the photocatalytic oxidation of pollutants with and without ferrate(VI). The pollutants examined were ammonia, cyanate (NCO−), formic acid (HCOOH), bisphenol-A (BPA), dibutyl phthalate (DBP), dimethyl phthalate (DMP) and microcystin-LR (MCLR). Some of these pollutants react sluggishly with either ferrate(VI) or UV-illuminated aqueous TiO2, and their oxidation could be enhanced in the presence of ferrate(VI). The role of ferrate(V) in enhancing the photocatalytic oxidation of pollutants is discussed. The review presents the fundamental approaches to enhance the photocatalytic oxidation of pollutants in water that can help to apply the process to real systems.
2 Reduction of ferrate(VI)
The photocatalytic reduction of ferrate(VI) in UV-irradiated aqueous TiO2 suspension has been performed in basic media as a function of TiO2 load (mass), ferrate(VI) concentration and pH (Sharma et al. 2001b). The photoreduction of ferrate(VI) in the TiO2 suspensions was faster than in the absence of TiO2. The photoreduction of ferrate(VI) to Fe(OH)3 in basic media can be expressed as Eq. 3.
The reaction of Fe(VI) to Fe(V) (Eq. F1 in Table 1) was postulated to be the rate-determining step because Fe(V) and Fe(IV) are unstable species and can be reduced by e −cb (Eqs. F2 and F3 in Table 1) at much faster rates than Fe(VI) (Menton and Bielski 1990; Rush et al. 1996). The photoreduction rate increased with TiO2 loading and gave a fractional order, 0.32 ± 0.04, with respect to Fe(VI) (Sharma et al. 2001b).
The initial rate of photocatalytic reduction under different conditions was analysed using a Langmuirian form (Eq. 4).
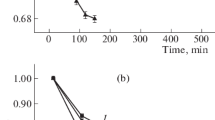
where k is the reaction rate constant and K is the apparent binding constant. The values of k decreased with increasing pH from 8.5 to 10.0 (k = 4.6–3.1 × 10−6 mol L−1 s−1 g−1 at 0.033 g TiO2 L−1 and k = 5.3–2.9 × 10−6 mol L−1 s−1 g−1 at 0.066 g TiO2 L−1). In this pH range, there are two ferrate(VI) species, monoprotonated HFeO4 − and deprotonated FeO 2−4 (pK a,HFeO4 = 7.23; Sharma et al. 2001c). The variation in the proportion of these two species with pH is shown in Fig. 1a. The decrease in reduction rates was determined to be related to the concentration of HFeO −4 species. This is demonstrated in Fig. 1b, which shows almost linear positive relationships between k and the fraction of HFeO −4 species at both TiO2 suspension loadings. An increase in electrostatic repulsion between the negatively charged TiO2 surfaces and the two ferrate(VI) species may occur with increase in the more negative FeO 2−4 species relative to HFeO −4 at higher pH values. This process will result in a slower photoreduction of ferrate(VI) at TiO2 surfaces at higher pH values.

a The variation in the fraction of ferrate(VI) species with pH. b Relationship between k and fraction of HFeO −4 species (α) at two TiO2 suspensions
3 Ammonia and cyanate
Initially, the photocatalytic reductions of ferrate(VI) in Fe(VI)–TiO2–UV–ammonia system under anoxic conditions at pH 9.0 under various concentrations of ferrate(VI) and ammonia were determined using 0.033 g L−1 TiO2 suspension and intensity (I) = 1.0 × 10−7 einstein s−1 (Sharma and Chenay 2005, 2008). A buffer solution consisting of phosphate and borate was used to maintain a solution pH of 9.0. The reduction rate of ferrate(VI) increased with increasing ferrate(VI) concentration at all ammonia concentrations, and the initial rate (R) may be expressed by Eq 5 as:
where \( \left[ {{\text{Fe}}\left( {{\text{VI}}} \right)} \right] = 118 - 990\mu {\text{M}},\left[ {{\text{Ammonia}}} \right] = 126 - 1044\mu {\text{M,}}\,a = 6.0 \times 10^{3} \mu {\text{M}}^{{0.25}} ,\,{\text{and}}\,{\text{b = 4}}{\text{.1}} \times {\text{10}}^{6} \mu {\text{M}}^{{1.25}} {\text{s}}^{{ - 1}} \)Next, the kinetic measurements of the photocatalytic reduction of ferrate(VI) in Fe(VI)–TiO2–UV–NCO− were carried out under anoxic conditions at pH 9.0 as a function of [NCO−], ferrate(VI), light intensity (I 0) and amount of TiO2 suspensions (Sharma et al. 2003; Winkelmann et al. 2008). The rate law can be expressed by Eq. 6:
where \( I_{0} = 6 \times 10^{{ - 8}} {\text{to1}}{\text{.5}} \times {\text{10}}^{{ - 6}} {\text{einstein}}\,{\text{L}}^{{ - 1}} {\text{s}}^{{ - 1}} ,\left[ {{\text{NCO}}^{ - } } \right] = 0.25 - 5.0 \times 10^{{ - 3}} {\text{mol}}\,{\text{L}}^{{ - 1}} \,{\text{and}}\,{\text{TiO}}_{2} = 0.03 - 0.1\,{\text{g}}\,{\text{L}}^{{ - 1}} . \) The oxidation of both ammonia and NCO− were found to be faster in the presence of ferrate(VI) than in the absence of ferrate(VI) in the solution mixtures (Fig. 2). In the case of ammonia, the increase in the rate of ammonia oxidation was related to the molar ratios of ferrate(VI) to ammonia (Sharma and Chenay 2005). An enhancement of the photocatalytic oxidation of ammonia and NCO− in the presence of ferrate(VI) is related to two processes: (1) inhibiting the h +vb –e −cb pair combination by reaction F1–F3 in Table 1, causing a greater amount of oxidant species, h +vb , O -•2 and •OH (reactions T1 and T3–T8, see Table 1) and (2) participation of highly reactive species, ferrate(V) and ferrate(IV) (reaction F2 and F3 in Table 1). The involvement of a second process is related to the reactivity of ferrate(V) and ferrate(IV) with ammonia and NCO-. The faster rates of such reactions than the rates of spontaneous decomposition of ferrate(V) and ferrate(IV) species (reactions F5 and F6 in Table 1) would indicate this possibility in enhancing the oxidation rates. The self-decomposition rates of ferrate(V) and ferrate(IV) are in the order of 106–107 M−1 s−1 in the alkaline pH range (Rush and Bielski 1994; Menton and Bielski 1990; Cabelli and Sharma 2008). In a recent study, the rate constant of ferrate(V) with NCO- was determined to be 9.6 × 102 M−1 s−1 at pH 10.9 and 22°C (Winkelmann et al. 2008), which is about four orders of magnitude slower than the reactions F2 and F3 in Table 1. Also, ferrate(IV) reacts even slower than ferrate(V); hence, the rate of ferrate(IV) reaction with NCO- would be much lower than 103 M−1 s−1. Based on this kinetic analysis, the participation of ferrate(V) and ferrate(IV) in enhancing the photocatalytic oxidation of NCO− in the presence of ferrate(VI) was ruled out. Hence, enhancement is most likely due to the first process involving the inhibition of h +vb and e −cb by ferrate(VI). The concentration of NCO− did not further decrease after about 2 h. This is related to a decrease in Fe(VI) concentration to a very low level after this time period, and hence, there is little beneficial effect of ferrate(VI) in the degradation of NCO−. A similar analysis concerning the oxidation of ammonia could not be carried out because the rate of ferrate(V) reaction with ammonia is not known at present. However, ferrate(V) has shown a high reactivity with amino compounds (Sharma and Bielski 1991; Bielski et al. 1994), which suggests the possibility of ferrate(V) involvement in enhancing the photocatalytic oxidation of ammonia in the presence of ferrate(VI) (Sharma and Chenay 2005).

The photocatalytic oxidation of ammonia (a) and cyanate (b) at pH 9.0. Experimental conditions: a TiO2 = 0.066 g L−1, [Ferrate(VI)] = 5.7 × 10−4 M, [Ammonia] = 9.4 × 10−4 M; b TiO2 = 0.60 g L−1, [Ferrate(VI)] = 5.0 × 10−4 M, [NCO−] = 1.0 × 10−3 M
4 Formic acid and bisphenol-A
The photocatalytic reduction of ferrate(VI) in the Fe(VI)–TiO2–UV–HCOOH reaction system at pH 9.0 has been determined under anoxic (deoxygenated) conditions as a function of ferrate(VI) concentration (100–970 μM) using 0.066 g L−1 TiO2 suspension (Sharma and Chenay 2008). The reduction rates of ferrate(VI) increased with increasing concentration of formic acid and were linear with [ferrate(VI)]. The initial reduction rate was expressed by Eq. 7:
Similar to the oxidation of ammonia and cyanate, the photocatalytic oxidation of HCOOH and BPA also showed an enhancement of their oxidation in the presence of ferrate(VI) (Sharma and Chenay 2008; Li and Li 2007). The rates of photocatalytic oxidation for both HCOOH and BPA increased with the following order of reaction systems: TiO2–UV–HCOOH (or BPA) < Fe(VI)–HCOOH (or BPA) < Fe(VI)–TiO2–UV–HCOOH (or BPA). In this study, it was found that not only the Fe(VI)–TiO2–UV system can enhance the BPA degradation but also significantly accelerate its further mineralisation in terms of dissolved organic carbon removal.
5 Dibutyl phthalate and dimethyl phthalate
The photocatalytic oxidation of DBP in Fe(VI)–TiO2–UV–DBP under deoxygenated conditions at pH 9.0 was examined (Li et al. 2008). DBP concentrations were determined using high-performance liquid chromatography (HPLC) with a high-pressure pump (Spectrasystem HPLC P4000), a UV detector (UV 6000LP) and an auto sampler (AS3000). In the HPLC analysis, a pinnacle II C18 column (5-μm particle size, 250 × 4.6-mm i.d.) was employed and a mobile phase of acetonitrile/water (80:20, v/v) was used at a flow rate of 1.0 mL/min. An injection volume of 20 μL was used and the concentration of DBP was determined by the UV detector at 227 nm. The results are shown in Fig. 3a and demonstrate that the concentration of DBP decreased faster than under either ferrate(VI) or TiO2–UV alone. The results clearly emphasise the role of ferrate(VI) in enhancing the oxidation of DBP. The effect of other oxidants, O2 and H2O2, on the photocatalytic oxidation of DBP was also examined and the results compared with ferrate(VI) in Fig. 3b. It should be pointed out that the oxidants were used separately and not in combination with ferrate(VI). It was evident that ferrate(VI) showed a greater oxidation effect than O2 and H2O2. Ferrate(VI) is a better electron acceptor than O2 [E 0 (Fe(VI)) = 0.72 V, E 0 (O2) = −0.13 V], and hence, it showed a greater enhancing effectiveness for the oxidation of DBP compared to O2. The results with H2O2 were unexpected in view of its superior reduction potential of 0.88 V compared to the other oxidants, but H2O2 gave the lowest enhancing effect. One reason may be that H2O2 is unstable in alkaline solution and rapidly decays into water and oxygen, and therefore, no expected enhancing effect of H2O2 could be seen.

The photocatalytic oxidation of DBP at pH 9.0 at UV intensity = 0.40 mW/cm2. a Degradation of DBP in different oxidation systems, [DBP] = 5–7 mg L−1, [Ferrate(VI)] = 0.08 mmol L−1, and TiO2 = 20 mg L−1. b Degradation of DBP in the presence of different electron acceptors, [DO] = 20 mg L−1, [H2O2] = 0.16 mmol L−1, [Ferrate(VI)] = 0.16 mmol L−1 and TiO2 = 20 mg L−1
The aqueous oxidation of DMP in a Fe(VI)–TiO2–UV–DMP system has been examined under deoxygenated conditions at pH 9.0 (Yuan et al. 2008a). The DMP concentration was determined using the same analytical procedure as described above for DBP, except that the mobile phase of acetonitrile/water (80:20, v/v) was used at a flow rate of 0.8 mL min−1 rather than 1.0 mL min−1. An injection volume of 20 μL was used and the concentration of DMP was determined by UV detection at 227 nm. The concentration of DMP decreased very slowly by either TiO2–UV illumination or ferrate(VI) alone, but decreased rapidly by photocatalysis in the presence of ferrate(VI) (Fig. 4a). Such a dramatic enhancement effect of ferrate(VI) is most likely due to the oxidation of DMP by the intermediate ferrate(V) and ferrate(IV) species, produced by the reduction of ferrate(VI) by e −cb (reactions F2 and F3, see Table 1). The inhibition of the hole–electron pair would not otherwise give such a rapid decrease in DMP in the Fe(VI)–TiO2–UV–DMP system.

The photocatalytic degradation of DMP under different conditions at pH 9.0 using TiO2 = 40 mg L−1, [ferrate(VI)] = 0.16 mmol L−1, [DMP]0 = 10.3 mg L−1and UV intensity = 0.40 mW cm−2. a Under N2 flow; b under different oxygen concentrations
The effect of oxygen concentration on the oxidation of DMP in the Fe(VI)–TiO2–UV–DMP system was also investigated (Yuan et al. 2008a). An increase in oxygen concentration decreased the photocatalytic oxidation efficiency, and no oxidation of DMP was seen in pure oxygen flow (see Fig. 4b). A postulation was made that the formation of an Fe–(organic) complex forms from the combination of reduced Fe(IV) or Fe(III) species, O2 and low concentration of DMP reaction products (Yuan et al. 2008a). Such a complex may be present in the bulk solution and adsorbed on the TiO2 surfaces. The adsorbed complexes reduce the adsorption of DMP on the TiO2 and thereby prevent the oxidation of DMP through h +vb interaction. Another possibility may also be considered for the diminishing effect of O2, which concerns the reactions F7–F10 (see Table 1) in the presence of O2. The increase in O2 level in the system would result in an increasing concentration of O -•2 and H2O2, which can react with reactive ferrate(V) species. This possibility would eliminate ferrate(V) without reacting with DMP. It appears that the rate of ferrate(V) reaction with O •-2 would be comparable to the rate for the reaction of ferrate(V) with DMP to give such an effect of oxygen concentration (see Fig. 4b). It should be pointed out that the photocatalytic oxidation of DMP in the presence of ferrate(VI) at the oxygen levels typically present in treated water may still exceed the performance of alternative chemical treatment methods (Yuan et al. 2008a). In the TiO2–UV–O2 system, the existence of •OH radicals was confirmed by electron spin resonance (ESR) spectroscopy upon irradiation at k = 355 nm. A 1:2:2:1 quartet (a N = a H = 1.49 mT) was observed upon irradiation. In the TiO2–Fe(VI) system without UV, a new but unknown radical (most likely an iron–oxo species) was believed to be formed and a septet spectrum was observed by the ESR spectroscopy (Yuan et al. 2008b).
6 Microcystin-LR
The photocatalytic oxidation of MCLR in the Fe(VI)-TiO2–UV–MCLR and Fe(III)–TiO2–UV–MCLR systems have been examined (Xing et al. 2002; Yuan et al. 2006). The results shown in Fig. 5a demonstrate that significant enhancement in the oxidation of MCLR was obtained in the presence of Fe(III) and ferrate(VI) in the system, and the effectiveness of ferrate(VI) was greater than that of Fe(III). Ferrate(VI) could achieve a degradation of almost 100% of MCLR in 30 min of contact time, and the degradation followed first-order kinetics (see Fig. 5b). The first-order rate constant, k ′, obtained for the ferrate(VI)–UV–TiO2 system was 2.5 and 4.4 times higher than for the Fe(III)–UV–TiO2 and UV–TiO2 systems, respectively.

a The photocatalytic degradation of MCLR. b Variation of Ln(CMCLR) versus time for the data given in a. Conditions: [ferrate(VI)] = 0.08 mmol L−1 and Fe(III) = 0.36 mmol L−1
The effect of five different ferrate(VI) dosages (0.04, 0.08, 0.13, 0.17 and 0.33 mmol L−1) was examined for the degradation of MCLR without controlling the pH, and the initial pH varied in the range of 6.0–7.0 (Yuan et al. 2006). As shown in Fig. 6a, the addition of ferrate(VI) increased the photocatalytic oxidation of MCLR at a contact time of 30 min, and a degradation of MCLR of up to 100% could be obtained for ferrate dosages of 0.08–0.17 mmol L−1. However, at the highest ferrate dosage of 0.33 mmol L−1, the degree of degradation reduced to 83%. Thus, a ferrate dose of 0.08 mmol L−1 was considered to be the optimum for the removal of MCLR. High concentrations of iron in the system could give detrimental effects by reducing the intensity of light to TiO2 surfaces and by creating cyclic reactions \( \left( {{\text{Fe}}^{\text{3 + }} {\text{ + e}}^{ - }_{\text{cb}} \to {\text{Fe}}^{\text{2 + }} \,{\text{and}}\,{\text{Fe}}^{\text{2 + }} {\text{ + h}}^{+}_{\text{vb}} \to {\text{Fe}}^{\text{3 + }} } \right) \). These reactions would not allow the oxidation of MCLR to proceed efficiently.

a Effect of ferrate(VI) concentration on the degree of photocatalytic degradation of MCLR. b The pseudo-first-order rate constant (k’, min−1) as a function of pH
The influence of pH on the photocatalytic oxidation of MCLR at a 0.13 mmol L−1 concentration was also investigated. At 30-min photocatalytic process, the removal efficiency of MCLR increased from 65% to 100% by increasing the pH from 2 to 6, but it decreased to 85% as pH increased further to 10.0 (Yuan et al. 2006). The variation of the first-order rate constant, k, obtained from the data collected at various pH values is shown in Fig. 6b. The k values increased from pH 2.0 to 6.0 and then decreased with pH. In highly acidic conditions, pH 2–4, the oxidation of MCLR probably occurs by way of free radical generation. The free radicals increase with pH, hence the increase in the degradation rate in the acidic pH range. Thus, it is speculated that ferrate(VI) was not participating to any significant degree in the removal of MCLR at pH 2.0–6.0. However, ferrate(VI) is increasingly stable in the pH range of 6.0 to 10.0 and must be involved in the oxidation of MCLR under these conditions. The decrease in rates in the pH range of 6.0–10.0 is related to an increase in electro-repulsion between TiO2 and ferrate(VI) species and a decrease in concentration of reactive HFeO −4 as discussed in Section 2. A nearly positive relationship between k and the fraction of HFeO −4 species (r 2 = 0.97) further suggests that the HFeO −4 species controls the oxidation of MCLR in the pH range of 6.0–10.0. Information from the application of HPLC analyses to the reaction between MCLR and ferrate(VI) at a dose of 0.08 mmol L−1 indicated changes to the Adda group and the opening/destruction of the heptapeptide ring of MCLR (Yuan et al. 2006). These findings are consistent with the measured reactivity of ferrate(VI) with amino acids of the MCLR (see Table 2), which also suggests that ferrate(VI) can effectively oxidise amino acids to detoxify MCLR (Sharma 2004).
7 Conclusions
The photocatalytic reduction of ferrate(VI) by UV-illuminated TiO2 suspension has been found to follow a Langmuirian form, and the reaction rate constant decreases with an increase in pH. The reactive ferrate(VI) species, HFeO −4 , was determined to be largely responsible for this pH dependence. The oxidation of pollutants in the Fe(VI)–TiO2–UV–pollutant system under anoxic conditions was found to be enhanced in comparison with ferrate(VI) or TiO2–UV alone. The combined effect of inhibiting e −cb –h +vb pair recombination and producing highly reactive ferrate(V) species may explain the observed enhancement of the oxidation. The role of ferrate(V) in the photocatalytic oxidation of pollutants in the presence of ferrate(VI) is determined by its reaction rate with the pollutants, which must be greater than the self-decomposition of ferrate(V) in order for there to be a significant enhancement. The enhancement by ferrate(VI) decreased when experiments were performed under air or O2 gas flow, and the reasons for this remain to be identified. Nevertheless, the oxidation performance of the Fe(VI)–TiO2–UV process in the treatment of aqueous pollutants is still considered superior to alternative chemical oxidation methods. Thus, a combination of ferrate(VI) and photocatalyst TiO2 can achieve the oxidation of recalcitrant pollutants in aqueous solutions.
8 Recommendations and perspectives
The formation of intermediate ferrate(V) species has been suggested in the photocatalytic reduction of ferrate(VI), but no direct evidence of this has been reported so far. It is believed that a method involving electron paramagnetic resonance spectroscopy could be applied to obtain direct evidence for the production of ferrate(V), since this technique was successfully applied to confirm the formation of Cr(V) in the heterogeneous photocatalytic reduction of Cr(VI) using TiO2 suspension in the presence of citrate and ethylenediaminetetraacetate (Testa et al. 2004; Meichtry et al. 2007). The rates for the reactivity of ferrate(V) with recalcitrant compounds such as BPA, DBP and DMP are required to fully assess the role of ferrate(V) in enhancing the photocatalytic oxidation of such compounds by ferrate(VI). All photocatalytic experiments using ferrate(VI) conducted so far used UV light as an illuminating source for Degussa TiO2 suspensions. This heterogeneous system is not efficient and studies are now emerging concerning the synthesis of modified TiO2 surfaces, which enable photocatalysis to occur under visible light wavelength irradiation; such a system would be more advantageous for practical applications. It is recommended that future experiments involving the photocatalytic oxidation of pollutants in the presence of ferrate(VI) should be performed under visible light conditions using modified TiO2 surfaces.
References
Bielski BHJ, Thomas MJ (1987) Studies of hypervalent iron in aqueous solutions. I. Radiation-induced reaction of iron(VI) to iron(V) by CO −2 . J Am Chem Soc 109:7761–7764
Bielski BHJ, Sharma VK, Czapski G (1994) Reactivity of ferrate(V) with carboxylic acids: a pre-mix pulse radiolysis study. Rad Phys Chem 44:479–484
Cabelli D, Sharma VK (2008) Aqueous ferrate(V) and ferrate(IV) in alkaline medium: generation and reactivity. ACS Symp Ser 985(Ferrates):158–166
Chae SY, Park MK, Lee SK, Kim TY, Kim SK, Lee WI (2003) Preparation of size-controlled TiO2 nanoparticles and derivation of optically transparent photocatalytic films. Chem Mater 15:3326–3331
Chen X, Mao SS (2007) Titanium dioxide nanomaterials: synthesis, properties, modifications, and applications. Chem Rev 107:2891–2959
Chenthamarakshan CR, Rajeshwar K, Wolfrum EJ (2000) Heterogeneous photocatalytic reduction of Cr(VI) in UV-irradiated titania suspensions: effect of protons, ammonium ions, and other interfacial aspects. Langmuir 16:2715–2721
Cozzoli PD, Comparelli R, Fanizza E, Curri ML, Agostiano A (2003) Photocatalytic activity of organic-capped anatase TiO2 nanocrystals in homogeneous organic solutions. Mater Sci Eng C C23:707–713
Dutta PK, Pehkonen SO, Sharma VK, Ray AK (2005) Photocatalytic oxidation of arsenic(III): evidence of hydroxyl radicals. Environ Sci Technol 39(6):1827–1834
Fujishima A, Honda K (1972) Electrochemical photolysis of water at a semiconductor electrode. Nature 238:37–38
Grätzel M (2001) Photoelectrochemical cells. Nature 414:338–344
Hoffmann MR, Martin ST, Choi W, Bahnemann DW (1995) Environmental applications of semiconductor photocatalysis. Environ Sci Technol 95:69–96
Hurum DC, Gray KA, Rajh T, Thurnauer MC (2005) Recombination pathways in the Degussa P25 formulation of TiO2: surface versus lattice mechanisms. J Phys Chem B 109:977–980
Jiang J-Q, Lloyd B (2002) Progress in the development and use of ferrate(VI) salt as an oxidant and coagulant for water and wastewater treatment. Water Res 36:1397–1408
Jiang J-Q (2007) Research progress in the use of ferrate(VI) for the environmental remediation. J Hazard Mater 146:617–623
Li C, Li XZ (2007) Degradation of endocrine disrupting chemicals in aqueous solution by interaction of photocatalytic oxidation and ferrate(VI) oxidation. Water Sci Technol 55(1–2):217–223
Li XZ, Yuan BL, Graham N (2008) Degradation of dibutyl phthalate in aqueous solution by a combined ferrate and photocatalytic oxidation process. ACS Symp Ser 985:364–377
Linesebigler AL, Lu G, Yates JT (1995) Photocatalysis on TiO2 surfaces: principles, mechanisms, and selected results. Chem Rev 95:735–758
Liu Z, He Y, Li F, Liu Y (2006) Photocatalytic treatment of RDX wastewater with nano-sized titanium dioxide. Environ Sci Pollut Res 13(5):328–332
Meichtry JM, Brusa M, Mailhot G, Grela MA, Litter MI (2007) Heterogeneous photocatalysis of Cr(VI) in the presence of citric acid over TiO2 particle; relevance of Cr(V)–citrate complexes. Appl Catal B: Environ 71:101–107
Menton JD, Bielski BHJ (1990) Studies of the kinetics, spectral and chemical properties of Fe(IV) pyrophosphate by pulse radiolysis. Radiat Phys Chem 36:725–736
Noorhasan NN, Sharma VK, Cabelli D (2008) Reactivity of ferrate (V) (FeVO 3−4 ) with aminopolycarboxylates in alkaline medium: a premix pulse radiolysis. Inorg Chim Acta 361:1041–1046
Prairie MR, Evans LR, Stange BM, Martinez SL (1993) An investigation of titanium dioxide photocatalysis for the treatment of water contaminated with metals and organic chemicals. Environ Sci Technol 27:1776–1782
Rush JD, Bielski BHJ (1994) Decay of ferrate(V) in neutral and acidic solutions. A premix pulse radiolysis study. Inorg Chem 33:5499–5502
Rush JD, Zhao Z, Bielski BHJ (1995) The oxidation of phenol by ferrate(VI) and ferrate(V): a pulse and stopped-flow study. Free Radic Res 22:349–360
Rush JD, Zhao Z, Bielski BHJ (1996) Reaction of ferrate(VI)/ferrate(V) with hydrogen peroxide and superoxide anion: a stopped-flow and premix pulse radiolysis study. Free Radic Res 24:187–198
Seluck H, Bekbolet M (2008) Photocatalytic and photoelectrocatalytic humic acid removal and selectivity of TiO2 coated photoanode. Chemosphere 73:854–858
Sharma VK, Bielski BHJ (1991) Reactivity of ferrate(VI) and ferrate(V) with amino acids. Inorg Chem 30:4306–4310
Sharma VK, O’Connor DB, Cabelli DE (2001a) Sequential one-electron reduction of Fe(V) to Fe(III) by cyanide in alkaline medium. J Phys Chem B 105:11529–11532
Sharma VK, Burnett CR, Rivera W, Joshi VN (2001b) Heterogeneous photocatalytic reduction of ferrate(VI) in UV-irradiated titania suspensions. Langmuir 17:4598–4601
Sharma VK, Burnett CR, Millero FJ (2001c) Dissociation constants of the monoprotic ferrate (VI) ion in NaCl media. Phys Chem Chem Phys 3:2059–2062
Sharma VK (2002a) Potassium ferrate (VI): an environmentally friendly oxidant. Adv Environ Res 6:143–156
Sharma VK (2002b) Fe(V) oxidation of pollutants: a premix pulse radiolysis study. Radiat Phys Chem 65:349–355
Sharma VK (2004) Use of iron(VI) and iron(V) in water and wastewater treatment. Water Sci Technol 49(4):69–74
Sharma VK, Burnett CR, O’Connor DB, Cabelli D (2002) Iron(VI) and iron(V) oxidation of thiocyanate. Environ Sci Technol 36(19):4182–4186
Sharma VK, Winkelmann K, Krasnova Y, Lee C, Sohn M (2003) Heterogeneous photocatalytic reduction of ferrate(VI) in UV-irradiated titania suspensions: role in enhancing destruction of nitrogen-containing pollutants. Int J Photoenergy 5(3):183–190
Sharma VK, Chenay BVN (2005) Heterogeneous photocatalytic reduction of Fe(VI) in UV-irradiated titania suspensions: effect of ammonia. J Appl Electrochem 35(7–8):775–781
Sharma VK, Burnett CR, Yngard RA, Cabelli DE (2005) Iron(VI) and iron(V) oxidation of copper(I) cyanide. Environ Sci Technol 39:3849–3854
Sharma VK, Mishra SK, Nesnas N (2006) Oxidation of sulfonamide antimicrobials by ferrate(VI) [FeVIO 2−4 ]. Environ Sci Technol 40:7222–7227
Sharma VK (2007) A review of disinfection performance of Fe(VI) in water and wastewater. Water Sci Technol 55(1–2):225–230
Sharma VK (2008) Oxidative transformations of environmental pharmaceuticals by Cl2, ClO2, O3, and Fe(VI): kinetics assessment. Chemosphere 73:1379–1386
Sharma VK, Chenay BVN (2008) Heterogeneous photocatalytic reduction of iron(VI): effect of ammonia and formic acid. ACS Symp Ser 985(Ferrates):350–363
Sharma VK, Yngard RA, Cabelli DE, Baum JC (2008) Ferrate(VI) and ferrate(V) oxidation of cyanide, thiocyanate, and copper(I) cyanide. Radiat Phys Chem 77(6):761–767
Testa JJ, Crela MA, Litter MI (2004) Heterogeneous photocatalytic reduction of chromium(VI) over TiO2 particles in the presence of oxalate: involvement of Cr(V) species. Environ Sci Technol 38:1589–1594
Winkelmann K, Sharma VK, Lin Y, Shreve KA, Winkelmann C, Hoisington LJ, Yngard RA (2008) Reduction of ferrate (VI) and oxidation of cyanate in a Fe(VI)–TiO2–UV–NCO−-system. Chemosphere 72(11):1694–1699
Xing H, Yuan BL, Wang Y, Qu J (2002) Photocatalytic detoxification of microcystins combined with ferrate pretreatment. J Environ Sci Health Part A 7:641–649
Yngard RA, Sharma VK, Filip J, Zboril R (2008) Ferrate (VI) oxidation of weak-acid dissociable cyanides. Environ Sci Technol 42:3005–3010
Yuan BL, Qu JH, Fu M-L (2002) Removal of cyanobacterial microcystis-LR by ferrate oxidation–coagulation. Toxicon 40(8):1129–1134
Yuan B, Li Y, Huang X, Liu H, Qu J (2006) Ferrate-assisted photocatalytic degradating of microcystin-LR using titanium dioxide. J Photochem Photobiol A Chem 178(1):106–111
Yuan BL, Li XZ, Graham N (2008a) Aqueous oxidation of dimethyl phthalate in a Fe(VI)–TiO2–UV reaction system. Water Res 42:1413–1420
Yuan BL, Li XZ, Graham N (2008b) Reaction pathways of dimethyl phthalate degradation in TiO2–UV–O2 and TiO2–UV–Fe(VI) systems. Chemosphere 72:197–204
Acknowledgements
The authors wish to acknowledge the support of the Florida Solar Energy program and the Hong Kong Government Research Grants Committee for some of the work described in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sharma, V.K., Graham, N.J.D., Li, XZ. et al. Ferrate(VI) enhanced photocatalytic oxidation of pollutants in aqueous TiO2 suspensions. Environ Sci Pollut Res 17, 453–461 (2010). https://doi.org/10.1007/s11356-009-0170-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-009-0170-0