
Abstract
Introduction
Severe acute malnutrition (SAM) is a major cause of child mortality worldwide, however the pathogenesis of SAM remains poorly understood. Recent studies have uncovered an altered gut microbiota composition in children with SAM, suggesting a role for microbes in the pathogenesis of malnutrition.
Objectives
To elucidate the metabolic consequences of SAM and whether these changes are associated with changes in gut microbiota composition.
Methods
We applied an untargeted multi-platform metabolomics approach [gas chromatography–mass spectrometry (GC-MS) and liquid chromatography–mass spectrometry (LC-MS)] to stool and plasma samples from 47 Nigerian children with SAM and 11 control children. The composition of the stool microbiota was assessed by 16S rRNA gene sequencing.
Results
The plasma metabolome discriminated children with SAM from controls, while no significant differences were observed in the microbial or small molecule composition of stool. The abundance of 585 features in plasma were significantly altered in malnourished children (Wilcoxon test, FDR corrected P < 0.1), representing approximately 15% of the metabolome. Consistent with previous studies, children with SAM exhibited a marked reduction in amino acids/dipeptides and phospholipids, and an increase in acylcarnitines. We also identified numerous metabolic perturbations which have not been reported previously, including increased disaccharides, truncated fibrinopeptides, angiotensin I, dihydroxybutyrate, lactate, and heme, and decreased bioactive lipids belonging to the eicosanoid and docosanoid family.
Conclusion
Our findings provide a deeper understanding of the metabolic consequences of malnutrition. Further research is required to determine if specific metabolites may guide improved management, and/or act as novel biomarkers for assessing response to treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Severe acute malnutrition (SAM) is a leading cause of global child mortality, is associated with growth faltering, and results in impaired cognitive development (Kar et al. 2008). A significant proportion of deaths due to numerous illnesses including malaria, measles and diarrheal diseases have also been attributed to malnutrition (Caulfield et al. 2004). SAM is most frequently defined by a weight-for-length/height Z-score (WHZ) < −3 and/or presence of oedema (WHO 2009). Middle upper arm circumference (MUAC) <11.5 cm is also commonly used in children aged 6-months (WHO 2009). Despite its prevalence, the metabolic changes that occur during malnutrition are poorly understood. A better understanding of these metabolic derangements may shed light on pathophysiology and/or adaptation to malnutrition, and inform improved clinical management.
Multiple studies have demonstrated amino acid depletion in children with SAM (Ghisolfi et al. 1978; Bartz et al. 2014), but untargeted studies pertaining to other metabolites are limited. Bartz et al. (2014) applied a targeted and untargeted approach to identify biomarkers associated with recovery and mortality in Ugandan children with SAM (Bartz et al. 2014). The concentrations of a number of metabolites were significantly altered upon rehabilitation, including amino acids, acylcarnitines and leptin. However, non-malnourished controls were not included, and the untargeted analysis was limited to gas chromatography–mass spectrometry (GC-MS) which covers only a small portion of the metabolome. Malnutrition in juvenile pigs has also revealed alterations in the metabolome, including changes in amino acids, choline metabolites, and products of microbial-mammalian co-metabolism (Jiang et al. 2015), suggesting the gut microbiota may be altered by malnutrition.
Differences in gut microbiota composition of children with SAM compared to healthy co-twins and unrelated controls have been described in Malawian and Bangladeshi children (Smith et al. 2013; Subramanian et al. 2014). Additionally, specific taxa capable of causing enteropathy when combined with a low protein diet have been identified in mice (Brown et al. 2015). Enteropathy resulting in malabsorption and increased intestinal permeability is associated with SAM in humans, as indicated by intestinal biopsies and dual sugar permeability tests (Denno et al. 2014). This phenomenon may be a more severe form of environmental enteric dysfunction (EED), which occurs almost universally in people exposed to poor hygiene and sanitation. The etiology of enteropathy in SAM is not well understood, but may involve repeated exposure to fecal enteropathogens combined with a low protein diet (Hashimoto et al. 2012; Brown et al. 2015).
The aim of this study was to identify conserved changes in the metabolome of children with SAM compared to controls in order to improve our understanding of the pathophysiology of malnutrition. Given the association with enteropathy and recent studies linking changes in the microbiota to SAM (Smith et al. 2013; Subramanian et al. 2014), we also measured biomarkers of intestinal inflammation and profiled the gut microbiota of these children.
2 Materials and methods
2.1 Study design and sample collection
We aimed to undertake a pragmatic study of unselected children admitted to a health facility in a region with endemic malnutrition to maximise the relevance of our findings to clinical practice. We recruited 58 children aged 6–48 months: 47 with SAM and 11 well-nourished hospital controls. Demographic and clinical characteristics at recruitment are shown in Table 1. Children aged 6–48 months with SAM (WHZ < −3 or MUAC <11.5 cms and/or nutritional oedema) admitted to the Federal Medical Centre, Gusau, Zamfara State, Nigeria between June 25th and September 21st, 2012 were invited to participate in the study. The hospital serves both urban and rural populations and recruitment occurred during the rainy season when malnutrition is most common. Written information about the study and a verbal explanation in the appropriate local language was provided to parents/carers and signed or thumb-printed consent secured. Clinical management was performed according to usual practice based on WHO guidelines including those for the management of SAM (WHO 2009). Well-nourished children (MUAC >12.5 cms or WHZ score ≥ −1 and no nutritional oedema) were recruited from the paediatric ward or outpatient clinics as controls during the same time period (Supplementary Table 1). HIV positive individuals were excluded from the study. In both groups, baseline demographic and clinical information was collected on standard forms by one of five clinicians trained in the research methods (Supplementary Table 1). The first available stool sample was collected and its consistency and the presence of visible blood and mucus were recorded. At the time of clinical sampling, approximately 2.5 mL of venous blood was collected into EDTA tubes for the purposes of the study. Ethical approval was provided by the Joint Ethical Review Committees of the University of Ibadan /University College Hospital, Ibadan, Nigeria.
2.2 Metabolite extraction from stool
Approximately 250 mg of wet stool was lyophilized overnight. After drying, 40 mg was weighed into microcentrifuge tubes and extracted with 8:2 methanol:H2O to a final concentration of 40 mg/mL. Samples were then vortexed for 30 s, followed by centrifugation for 15 min at 10,000 rpm. 50 μL of this supernatant was dried in a speedvac for GC-MS analysis and the remaining stored at −80 °C for LC-MS.
2.3 Metabolite extraction from plasma
Metabolites were extracted from plasma according to the methods of Dunn et al. 2011. Briefly, plasma samples were thawed on ice for 30 min. Once thawed, 805 μL of 8:2 methanol:H2O was added to 230 μL of plasma to make a 4.5 fold dilution. Samples were vortexed for 15 s and centrifuged at 15,000 rpm for 15 min to pellet precipitated proteins. 370 μL of supernatant was then transferred to separate vials and dried down for GC-MS and LC-MS using a speedvac with no heat.
2.4 Untargeted GC-MS analyses
For stool, samples were derivatized with 40 μL of 2% methoxyamine-HCl in pyridine (MOX) incubated at 50 °C for 90 min, followed by 20 μL of N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) for 30 min at 50 °C. Samples were then transferred to micro-inserts before analysis by GC-MS. For plasma, 50 μL of MOX was added to dried samples and incubated for 90 min at 50 °C. 50 μL of MSTFA was then added and incubated for 30 min at 50 °C. Samples were then transferred to 1.5 ml microcentrifuge tubes and centrifuged at 15,000 rpm for 5 min to pellet debris. This supernatant was then transferred to HPLC vials containing inserts for analysis.
One μL of sample was injected into an Agilent 7890A GC, 5975 inert MSD with triple axis detector. Samples were injected using pulsed splitless mode using a 30 m DB5-MS column with 10 m duraguard, diameter 0.35 mm, thickness 0.25 μm (J&W Scientific, Folsom, USA). Helium was used as the carrier gas at a constant flow rate of 1 mL/min. Oven temperature was held at 70 °C for 5 min then increased at a rate of 5 °C/min to 300 °C and held for 10 min. Solvent delay was set to 7 min, and total run time was 61 min. Masses between 25 and 600 m/z were selected by the detector. All samples were run in random order and a single sample was run with every batch as a quality control to ensure machine consistency.
Chromatogram files were deconvoluted and converted to ELU format using AMDIS Mass Spectrometry software (Stein 1999) with the sensitivity set to low, resolution to medium, and support threshold to high. Chromatograms were aligned using Spectconnect (http://spectconnect.mit.edu) (Styczynski et al. 2007) with the support threshold set to low. The integrated signal (IS) matrix output was used for all further analysis. Zeros were replaced with two-thirds the minimum detected value on a per metabolite basis (Timotej 2011), followed by a log base 2 transformation. All further analyses were performed using these log-transformed values.
Metabolites were initially identified by comparison to the NIST 11 standard reference database (http://www.nist.gov/srd/nist1a.cfm). Identities of metabolites of interest were then confirmed by authentic standards if available.
2.5 Untargeted LC-MS analyses
For stool, 2.5 μL of 1 μg/mL 13C6 phenylalanine internal standard (Cambridge Isotopes, Tewksbury, USA) was added to 47.5 μL of extracted stool. For plasma, dried samples were reconstituted in 85.5 μL of ddH2O. 4.5 μL of 1 μg/mL 13C6 phenylalanine in ddH2O was then added to each vial as an internal standard. Samples were vortexed for 15 s, then transferred to microinserts and injected into an Agilent 1290 Infinity HPLC coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, USA) with a HESI (heated electrospray ionization) source. For HPLC, 2 μL of each sample was injected into a ZORBAX Eclipse plus C18 2.1 × 50 mm × 1.8 micron column. Mobile phase (A) consisted of 0.1% formic acid in water and mobile phase (B) consisted of 0.1% formic acid in acetonitrile. The initial composition of 0% (B) was held constant for 30 s and increased to 100% over 3.0 min. For stool, mobile phase B was held at 100% for 1 min and returned to 0% over 30 s for a total run time of 5 min. For plasma, mobile phase B was held at 100% for 2 min and returned to 0% over 30 s for a total run time of 6 min.
Full MS scanning between the ranges of m/z 50–750 was performed on all samples in both positive and negative mode at 140,000 resolutions. The HESI source was operated under the following conditions: nitrogen flow of 30 and 8 arbitrary units for the sheath and auxiliary gas respectively, probe temperature and capillary temperature of 450 °C and 250 °C respectively and spray voltage of 3.9 and 3.5 kV in positive and negative mode respectively. The automatic gain control (AGC) target and maximum injection time were 1e6 and 500 ms respectively. For molecular characterization, every tenth sample was also analyzed with a data dependent MS/MS method where a 35,000 resolution full MS scan identified the top 12 signals above a 1e5 threshold which were subsequently selected at a 1.2 m/z isolation window for MS/MS. Normalized collision energy for MS/MS was 28, resolution 17,500, AGC target 1E5 and maximum injection time was 60 ms. Blanks of pure methanol were run between every sample to limit carryover, and a single sample was run multiple times with every batch to account for any machine inconsistency. After data acquisition Thermo .RAW files were converted to .MZML format and centroided using ProteoWizard (Kessner et al. 2008). Files were then imported into R using the XCMS package (Patti et al. 2012) for chromatogram alignment and deconvolution. Features were detected with the “xcmsSet” function using the “centWave” method and a ppm tolerance of 1. Prefilter was set to 3–5000, noise 1E5, and signal to noise threshold was set to 5. Due to a lower overall noise and signal in negative mode, noise was set to 1E3 for this mode. Retention time correction was conducted using the “obiwarp” method, grouping included features present in at least 25% of all samples, allowable retention time deviation was 5 s, and m/z width set to 0.015. Areas of features below the signal to noise threshold in the data were integrated using the “fillPeaks” function with default settings. Any remaining zeros in the data were then replaced with two-thirds the minimum value on a per mass basis (Timotej 2011) before log base 2 transformation. The log-transformed mass list was then exported as a single .txt file and used for all further analyses. All further analyses were carried out in R unless otherwise specified. Positive and negative mode data were treated as two independent datasets for all analyses.
Metabolites were putatively identified based on accurate mass and LC-MS/MS fragmentation patterns (Supplemental Fig. 1). Predictions were made mainly by de novo compound identification from in depth investigation of individual MS/MS spectra, utilizing the METLIN database (http://metlin.scripps.edu) (Smith et al. 2005), Human Metabolome Database (http://www.hmdb.ca) (Wishart et al. 2007), and CFM-ID (http://cfmid.wishartlab.com) (Allen et al. 2014) whenever possible to aid in identification. Metabolites of interest were confirmed by authentic standards when available based on accurate mass, retention time and MS/MS spectra (Supplemental Table 2).
2.6 Statistical analysis of metabolome data
Principal Component Analysis (PCA) was conducted in R using the “FactoMineR” package with pareto scaling. Components 1 and 2, representing approximately 15% and 9% of the variation respectively, could not be explained by any of the metadata collected (Supplemental Fig. 2). We therefore selected the lowest components which maximized separation between SAM and controls (components 3 and 4) for qualitative analysis of the metabolome. Partial Least Squares Discriminant Analysis (PLS-DA) models were constructed in SIMCA 13.0.3.0 (Umetrics) using pareto scaling and sevenfold cross-validation. The number of components was selected using the autofit function which selects the maximum number of significant components according to cross-validation rules. Significant models were defined as those with cross-validated analysis of variance (CV-ANOVA) P values less than 0.05. Metabolites that differed between SAM and controls were determined independently of PCA analysis using unpaired Wilcoxon tests with Benjamini-Hochberg [False Discovery Rate (FDR)] corrections to account for multiple hypothesis testing (Benjamini and Hochberg 1995). Metabolites with a corrected P < 0.1 and average fold change >2 in either group (SAM or control) were selected for further investigation. Fold changes were calculated using the geometric mean to limit inflation of fold change values due to outliers. Boxplots and stripcharts were constructed in R using the ggplot2 package. Odds ratios of metabolites to identify SAM from controls were calculated from conditional logistic regressions performed on all metabolites using the glm function in R with 10,000 iterations and a binomial distribution. Receiver operating characteristic (ROC) curves and forest plots were built in R using the pROC and forestplot packages respectively. In the case of Des ADS-FPA, the 13C isotope was used for statistical analyses and plotting due to a large singly-charged interfering peak with mass similar to the 12C Des ADS-FPA. All raw data files were manually inspected to ensure the data was accurately represented by the 13C peak. The effect of potential confounding variables was investigated using Spearman’s correlations for continuous variables and Wilcoxon or Kruskal Wallace tests for discrete variables. Tests with FDR corrected P values less than 0.1 were considered significant.
2.7 Calprotectin and lactoferrin stool ELISAs
Calprotectin was measured using the IDK® Calprotectin ELISA Kit along with the IDK Extract® stool extraction kit (Immundiagnostik, Bensheim, Germany). Lactoferrin was measured using the Lactoferrin Scan™ ELISA kit (Techlab®, Blacksburg, USA). All protocols were followed as per the manufacturer’s instructions.
2.8 Microbiome profiling
DNA was extracted from stool samples using the PowerSoil-htp 96 Well Soil DNA isolation kit from MoBio (Carlsbad, CA) according to the manufacturer’s protocol, with modifications as outlined by the Earth Microbiome Project (version 4_13). Approximately 250 mg of fecal sample was used for the extractions. Samples were sequenced by amplifying the V4 hypervariable region of the 16S rRNA gene. Sample amplification for sequencing was carried out using the forward primer (ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNN(8)GTGCCAGCMGCCGCGGTAA) and the reverse primer (CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTNNNN(8)GGACTACHVGGGTWTCTAAT) where nnnn indicates four randomly incorporated nucleotides, and (8) was a sample-specific nucleotide barcode where the barcodes differed by an edit distance of at least 4. The 5′ end is the adapter sequence for the Illumina MiSeq sequencer and the sequences following the barcode are complementary to the V4 rRNA gene region. Amplification was carried out in 42 μL with each primer present at 3.2 pmol/μL, 20 μL GoTaq hot start colorless master mix (Promega) and 2 μL extracted DNA. The PCR protocol was as follows: initial activation step at 95 °C for 2 min and 25 cycles of 1 min 95 °C, 1 min 50 °C and 1 min 72 °C.
All subsequent work was carried out at the London Regional Genomics Centre (LRGC, lrgc.ca, London, Ontario, Canada). Briefly, PCR products were quantified with a Qubit 2.0 Flourometer and the high sensitivity dsDNA specific fluorescent probes (Life Technologies). Samples were mixed at equimolar concentrations and purified with the QIAquick PCR Purification kit (QIAGEN). Samples were paired-end sequenced on an Illumina Mi-Seq with the 600 cycle version 3 reagents with 2 × 220 cycles.
Resulting reads were extracted and de-multiplexed using modifications of in-house Perl and UNIX-shell scripts with operational taxonomic units (OTUs) clustered at 97% identity, similar to our reported protocol (Gloor et al. 2010). Automated taxonomic assignments were carried out by comparison to the SILVA database (http://www.arb-silva.de/). Supplemental Table 1 displays the nucleotide barcodes and their corresponding samples. To control for background contaminating sequences, a no-template control was also sequenced. Barplots were constructed with R (r-project.org) using proportional values. Rare OTUs found at less than 1% abundance in all samples were grouped in the remainder for barplots only. All other analyses were conducted with all OTUs. Principal Coordinate Analysis (PCoA) plots were constructed in Qiime (Caporaso et al. 2010) (qiime.org) using weighted UniFrac distances.
To avoid inappropriate statistical inferences made from compositional data, centred log-ratios (clr), a method previously described by Aitchison (Aitchison 1982), and adapted to microbiome data was used with unpaired Wilcoxon tests for comparisons of OTU level data (Fernandes et al. 2013, 2014). The Benjamini Hochberg (FDR) method was used to control for multiple testing with a significance threshold of 0.1. All statistical analysis, unless otherwise indicated, was carried out using R (r-project.org).
3 Results
3.1 Study population
The demographics of the study population is displayed in Table 1. Amongst the 58 children recruited, 47 were diagnosed with SAM (WHZ < −3 and/or MUAC <11.5 and/or oedema), of which 26 (55.3%) were diagnosed with kwashiorkor. Apathy was present in 34 (72.3%), anorexia in 42 (89.4%), thin hair in 42 (89.4%), glossitis in 27 (57.4%), one or more Bitot spots in 9 (19.1%), dermatitis in 22 (46.8%), stomatitis in 27 (57.4%) and 18 (38.3%) had oral ulceration. Abdominal distension was present in 11 (23.4%), hepatomegaly in 28 (59.6%) and splenomegaly in 26 (55.3%). Loose or watery stools (Bristol Stool Form Scale 5–7) occurred in 36 (76.6%) of the SAM cases compared with 4 (36.4%) controls (Table 1). None of the children had bloody stools. The distribution of ethnicity and area of residence differed significantly between cases and controls.
3.2 The fecal microbiota of Nigerian children
To determine if the composition of the gut microbiota differed between Nigerian children with SAM and controls, we profiled the microbiota of stool samples by amplifying the V4 region of the 16S rRNA gene. Two samples did not have enough material for DNA extraction, leaving 45 SAM and 11 controls remaining. The fecal microbiota was dominated by Enterobacteraciae, Bifidobacterium, Enterococcus, Pediococcus, Lactobacillus and Streptococcus species (Fig. 1a). There were no differences in alpha or beta diversity between SAM and controls (Fig. 1b, c), nor were there any individual taxa that differed significantly between groups (Wilcoxon test, FDR corrected P > 0.1, Supplementary Table 3). Although there was a trend for lower bacterial diversity in children with SAM compared to controls, the difference was not significant, even when age was taken into account (Fig. 1c, ANCOVA P > 0.05).

The stool microbiota does not discriminate SAM from controls. a Stool microbiota profiled using the V4 region of the 16S rRNA gene. Each bar in the barplot represents a single sample from a single child and each color a different bacterial taxa (OTU). OTUs present at less than one percent in every sample were placed in the remainder displayed in black. Samples are ordered by their WHZ displayed in the heatmap above. b Principal Coordinate Analysis (PCoA) plot of microbiota profiles built from weighted UniFrac distances. Each point represents a single sample from a single child. Positions of points display dissimilarities in the microbiota, with points further from one another being more dissimilar. c Shannon’s diversity of the stool microbiota in children with SAM compared with controls. The boxes represent the 25th and 75th quartiles, and the line displays the median value within each group. Points extending beyond the lines are outliers defined as values greater or less than 1.5 times the interquartile range
3.3 Fecal metabolome and inflammatory markers do not distinguish SAM from controls
To obtain a global view of nutrient deficiency in SAM and gain insight into pathophysiology, we performed a comprehensive untargeted analysis of metabolites in stool from all 58 children using two different methods; gas chromatography–mass spectrometry (GC-MS) and liquid chromatography–mass spectrometry (LC-MS). Surprisingly, both methods yielded no metabolites significantly affected by SAM (Wilcoxon test, FDR corrected P > 0.1, Supplementary Fig. 3; Supplementary Table 4). There was no separation of groups based on qualitative PCA analysis, and PLS-DA models had low predictability and were not significant (Positive ESI model R2 = 0.46, Q2 = 0.08, CV-ANOVA p = 0.085,1 component) (Supplementary Fig. 4). There were also no significant differences in the inflammatory proteins calprotectin or lactoferrin, as measured by ELISA (Supplementary Fig. 5).
3.4 Children with SAM have a distinct plasma metabolome
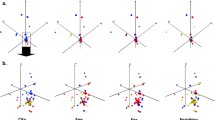
In contrast to stool results, the abundance of approximately 15% of LC-MS features (defined as a unique m/z and retention time) detected in plasma were significantly altered by SAM (Wilcoxon test, FDR corrected P < 0.1, Supplementary Table 5). Moderate separation between groups was observed along the 3rd and 4th component of principal component analysis (PCA) plots built from LC-MS data (Fig. 2, Supplementary Fig. 2 shows principal components 1 and 2), demonstrating the plasma metabolome of children with SAM is distinct from non-malnourished children. To further validate these findings, supervised PLS-DA models were constructed from positive ESI data (Supplementary Fig. 6). The PLS-DA model showed moderate predictability and was significant (R2 = 0.98, Q2 = 0.69, CV-ANOVA p = 1.43E−05, 5 components), indicating that the plasma metabolome of children with SAM was unique from healthy controls. The vast majority of differential features were detected exclusively by LC-MS, with only three metabolites differing significantly by GC-MS (valine, leucine/isoleucine and aspartic acid). There were no metabolites that differed between children with non-oedematous malnutrition (marasmus, N = 21) or oedematous malnutrition (kwashiorkor, N = 26), and no significant effect of sex, age, formula feeding, or residence was observed (data not shown).

Principal component analysis (PCA) plots of plasma metabolome from children with SAM and controls as determined by LC-MS using positive (top) or negative (bottom) electrospray ionization (ESI). Only components 3 and 4 are shown. The plots on the left display individual samples (scores). Each point represents a single sample from a single child. Positions of points display dissimilarities in the metabolome, with points furthest from one another being most dissimilar. Plots on the right display individual LC-MS features (loadings). Each ray represents a single LC-MS feature, with those significantly elevated in SAM or Controls highlighted in black or red respectively (Wilcoxon test, FDR corrected P < 0.1)
3.5 Oxylipins, phospholipids and amino acids are depleted in the plasma of children with SAM
Significant features in LC-MS data with greater than twofold change between SAM and controls were selected for further identification. As expected, free amino acids and dipeptides were lower in children with SAM including glutamine, arginine, tyrosine, leucine/isoleucine, valine, and the tryptophan metabolite kynurenine (Fig. 3a). A number of ether-linked single chain phospholipids belonging to the phosphatidylcholine (PC) and phosphatidylethanolamine (PE) families were also significantly decreased (Fig. 3b). Additionally, there was a marked reduction in a number of oxylipins belonging to the eicosanoid and docosanoid family. In depth investigation of fragmentation patterns of these lipids revealed that each feature (represented as a single boxplot pair in Fig. 3c) contained at least two different oxylipin species, differing only in the location of the hydroxyl group(s). The precise species contributing to the differences between SAM and controls could therefore not be determined. Arachidonic acid (AA), a precursor to the eicosanoids was also significantly lower in children with SAM. While median abundance of docosahexaenoic acid (DHA, the precursor to the docosanoids), was lower in children with SAM, the difference was not significant (FDR corrected P = 0.2) (Fig. 3c).

Free amino acids/dipeptides (a), phospholipids (b), and oxylipins (c) significantly decreased in children with SAM compared to controls (>twofold change, Wilcoxon test, FDR corrected P < 0.1, with the exception of DHA which was not significant). Panels a, b were detected by positive ESI LC-MS and c by negative ESI LC-MS. Each point represents a single sample from a single child. The boxes represent the 25th and 75th quartiles, and the line displays the median value within each group. Points extending beyond the lines are outliers defined as values greater or less than 1.5 times the interquartile range. PC phosphatidylcholine, PE phosphatidylethanolamine. See Supplementary Table 5 for phospholipid identities. *Metabolite ID confirmed by authentic standards
3.6 Metabolites elevated in plasma of children with SAM
Unlike metabolites negatively associated with SAM, the classes of metabolites elevated in malnourished children were more diverse, and included sugars, peptides, lipids, short chain fatty acids and porphyrins among others. Of interest, we detected a dihexose (a disaccharide composed of two six carbon hexoses) by LC-MS in both positive and negative ionization modes that was significantly elevated in children with SAM (Wilcoxon test, FDR corrected P < 0.1, Fig. 4). Authentic standards of different dihexoses confirmed that our LC-MS measurements represented the total dihexoses in plasma. Analysis of standards by GC-MS, which is able to resolve dihexoses, and LC-MS/MS determined that sucrose was the most abundant dihexose in plasma, with smaller amounts of lactose and maltose also present.

Metabolites significantly elevated at least twofold in the plasma of children with SAM compared to controls (Wilcoxon test, FDR corrected P < 0.1). Left Metabolites detected using positive ESI LC-MS. Right Metabolites detected using negative ESI LC-MS. Each point represents a single sample from a single child. The boxes represent the 25th and 75th quartiles, and the line displays the median value within each group. Points extending beyond the lines are outliers defined as values greater or less than 1.5 times the interquartile range. FPA fibrinopeptide A, HTD 3-hydroxy-cis-5-tetradecenoyl. *Metabolite ID confirmed by authentic standards
Two forms of truncated fibrinopeptide A (FPA) missing 2, or 3 N-terminal amino acids were also detected in significantly greater amounts in children with SAM. Specifically, des-AD (lacking N-terminal alanine and aspartate) and des-ADS FPA (lacking N-terminal alanine, aspartate and serine) were both increased approximately sixfold (Fig. 4). Intact FPA was outside the mass range of our initial analysis, and therefore we conducted a second analysis with a larger mass range to include the intact FPA peptide. Surprisingly, the abundance of intact FPA was not significantly different between SAM and controls (Supplementary Fig. 7), indicating that the truncated forms are not simply a degradation product of increased intact FPA, but have some other unknown origin.
Other metabolites of interest elevated in SAM included several acylcarnitines, the peptide hormone angiotensin I (Ang I), heme, lactate, oleoyl ethanolamide, 2,4- and 3,4-dihyroxybutyrate, an uncharacterized sphingoid base, a hydroxyvitamin D3 derivative and several other unknown compounds (Fig. 4; Supplementary Table 5).
3.7 Assessment of metabolites to discriminate SAM from controls
To measure the strength of the association between metabolites and SAM, we calculated odds ratios (OR) based on conditional logistic regressions of all metabolites elevated in children with SAM (Supplementary Table 6). Dihexoses and lactate were among the metabolites with the highest OR, ranging from 2.25 to 2.20 respectively (Fig. 5a). ROC curves identified decanoylcarnitine, dihexoses, an uncharacterized sphingoid base, angiotensin I, and heme as the metabolites that maximized the sensitivity and specificity for SAM, as shown by the area under the curve (AUC), which ranged from 0.83 to 0.81 for these compounds (Fig. 5b; Supplementary Table 6).

Evaluation of metabolites to identify SAM from controls. a Odds Ratios as determined by conditional logistic regressions of all validated metabolites positively associated with SAM. Bars represent 95% confidence intervals. b Receiver Operator Characteristics (ROC) curves. Metabolites with the highest area under the curve (AUC) are shown
4 Discussion
We report the first LC-MS-based untargeted metabolomic study of stool and plasma from children with SAM. In contrast to the stool microbiota and metabolome, which did not discriminate SAM from controls, approximately 15% of the plasma metabolome, equating to 585 features, were significantly altered in malnutrition. The explanation for the lack of differences in stool are likely multifaceted, but may include the large effect of diet and inter-individual variation which cannot be accounted for with small cross-sectional studies. It is also worth noting that feces represent the net result of nutrient consumption, digestion and absorption. Malabsorption has been widely reported in SAM (Kvissberg et al. 2016), and therefore it is possible that these children both consume and absorb fewer nutrients compared with non-malnourished controls, resulting in little net difference in stool nutrient composition.
The stool microbiota data mirrored the metabolome results as neither the composition nor diversity were significantly altered by malnutrition, even when children were matched for age. This is in contrast to previous studies that have observed differences in the relative abundance of specific taxa and diversity for age in twin pairs and unrelated controls (Smith et al. 2013; Subramanian et al. 2014). The relatively small number of controls in our study limited our ability to model the relationship between microbial diversity and age, and therefore it is possible that a larger sample size may have revealed significant differences. Our cohort also tended to be older than the children included in the study by Subramanian et al. (Subramanian et al. 2014), which could also partially explain the lack of differences in the microbiota. As the microbiota has been shown to stabilize by around 2 years of age (Subramanian et al. 2014), and a large proportion (41%) of the children in our study were 2 years or older, it is possible that the window where microbiota stunting is most apparent was not captured by our study. However, none of the plasma metabolites affected by SAM were of clear microbial origin, providing further evidence that the microbiota may not be playing a major role in this particular cohort.
Among the plasma metabolites discriminating SAM from controls, the total dihexoses had one of the strongest positive associations, as determined by ROC and OR analyses, and were detected in both positive and negative ionization modes. Regardless of their structure and content in the diet, disaccharides are not readily absorbed, and therefore must pass through the intercellular space of the intestinal mucosa to reach systemic circulation. For this reason, sucrose, cellobiose, lactulose and other disaccharides have been used as indicators of intestinal permeability (Pearson et al. 1982; Sutherland et al. 1994). Malabsorption and increased intestinal permeability are associated with SAM and EED, as measured by dual sugar permeability tests such as the lactulose/mannitol test (Denno et al. 2014; Kvissberg et al. 2016). Although these tests are fairly reliable, the requirement for fasting prior to administration raises ethical concerns for children with SAM, and urine must be collected over several hours for accurate results. A non-administered permeability test, for which the total dihexoses are an attractive candidate, would therefore be highly valuable.
Although elevated dihexoses are suggestive of enteropathy in the SAM group, we did not observe any difference in the inflammatory markers calprotectin or lactoferrin in stool. These proteins are used as indicators of inflammation in other pathologies such as inflammatory bowel disease (Smith and Gaya 2012), but have not been evaluated as biomarkers of enteropathy in SAM. The weak correlation between lactoferrin and calprotectin (Spearman’s R = 0.33, P = 0.01) limits our ability to make conclusions as to the intestinal inflammatory status of these children. Future studies evaluating the association of non-administered disaccharides with enteropathy are warranted to determine if they might also be useful in identifying EED.
The majority of metabolites discriminating SAM from controls in plasma were consistent with nutritional depletion due to malnutrition. As expected, a number of amino acids/dipeptides were reduced, indicative of protein deficiency. Oxylipins were also significantly decreased, and to the best of our knowledge, have not been examined in children with SAM previously. Importantly, each oxylipin feature contained at least two different species, and therefore the number and diversity of oxylipins altered by SAM may be vastly under-represented by our study. Oxylipins are bioactive lipids formed by oxidation of long chain polyunsaturated fatty acids (LCPUFA), with the most well studied being the AA-derived eicosanoids (Gabbs et al. 2015). These bioactive lipids perform a wide array of functions, including tissue repair, blood clotting, and regulation of the immune system. Oxylipins within the same family can have similar or opposing effects (i.e. pro- or anti-inflammatory), and therefore we cannot determine the precise biological consequence of reduced oxylipins during SAM (Gabbs et al. 2015). However, as children with SAM exhibit an impaired immune response to a variety of pathogens (Schaible and Kaufmann 2007), a lack of oxylipin mediators may be a contributing factor and warrants further investigation.
Previous studies have confirmed that children with SAM are deficient in LCPUFA (Holman et al. 1981; Leichsenring et al. 1995), including the oxylipin precursors AA and DHA. A large proportion of LCPUFA are stored as acyl-linked phospholipids, which could not detectable by our method, and therefore a comparison of the total LCPUFA in our cohort was not possible. Nevertheless, the significant reduction in free AA, non-significant trend for lower free DHA, and significant decreases in multiple phospholipids and oxylipins are suggestive of LCPUFA deficiency in our cohort.
Despite previous evidence of LCPUFA deficiency in SAM, the levels of LCPUFA in ready-to-use therapeutic food (RUTF) are low (Jones et al. 2015). Recently, a RUTF formulation supplemented with fish oil has been developed, which translated into increased LCPUFA in recipient children (Jones et al. 2015). More studies are required to determine if the incorporation of fish oil into refeeding programs restores oxylipin levels as well and whether this leads to any clinical benefit.
Apart from dietary deficiencies, other factors may also influence LCPUFA and oxylipin levels. Children with SAM have decreased desaturase activity, resulting in decreased synthesis of AA and DHA from their precursors (Koletzko et al. 1986; Smit et al. 2004). Additionally, a large proportion of dietary fatty acids are lost to beta-oxidation, a process that is induced during starvation (Nagao et al. 1993; Cunnane and Anderson 1997). Interestingly, a number of even-chain acylcarnitines, the bi-products of beta oxidation, were significantly elevated in children with SAM in our study. This suggests beta-oxidation of lipids may contribute in part to LCPUFA deficiency in SAM. Bartz et al. 2014 found that even-chain acylcarnitines, including the C10, C8 and C2 species identified in our study, decreased significantly upon nutritional intervention in children with SAM from Uganda. Acylcarnitines may therefore be useful not only as biomarkers of a malnourished state, but also as measures of treatment efficacy.
The truncated FPAs were also metabolites of interest as they are unique peptides of unknown origin and were elevated approximately sixfold in SAM. The des-ADS form in particular has not been reported previously. FPA is a 16 amino acid peptide produced upon cleavage of the fibrinogen alpha chain by thrombin during the coagulation cascade. Thus it is an indicator of thrombosis, and is elevated in plasma during a number of inflammatory conditions including Crohn’s disease, gastric cancer, and coronary thrombosis (Eisenberg et al. 1985; Edwards et al. 1987; Ebert et al. 2006). As the intact peptide was not elevated in SAM, these fragments are not simply a result of increased inflammation and thrombosis, but have another unknown origin. One hypothesis is that they may arise from increased protease activity as proposed by Zhang et al. (2013) who reported elevated des-A FPA in gastric cancer with lymph node metastasis. Protease activity is increased during starvation to supply the body with additional amino acids (Medina et al. 1991), and therefore it is possible that these truncated forms are a consequence of this up-regulation.
Lactate was also elevated in the plasma of malnourished children, and was one the metabolites with the highest OR for SAM. Lactate is an endproduct of cellular respiration under anaerobic conditions, and is elevated in blood due to a number of etiologies, including anemia, sepsis, trauma, and malignancy (Reddy et al. 2015). In the context of malnutrition, elevated lactate may result from insufficient oxygen supply due to the anemia or increased infections associated with SAM. Interestingly, several studies have shown blood lactate to be a good predictor of mortality due to a variety of illnesses (Krishna et al. 1994; Shapiro et al. 2005; Mikkelsen et al. 2009), including a study of Tanzanian children with any febrile illness (Mtove et al. 2011). Further research should evaluate lactate as a predictor of mortality in SAM.
Finally, we observed a significant increase in free iron(III) heme in the plasma of malnourished children. Heme is normally bound to hemeproteins, but is released under conditions of oxidative stress and as a result of hemolysis. Free heme causes tissue damage, systemic inflammation (Balla et al. 1991; Wagener et al. 2001), and exacerbates sepsis and malaria in animal models (Seixas et al. 2009; Larsen et al. 2010). In humans, malaria severity correlates with the levels of free heme, and sepsis-related mortality is associated with decreased levels of the heme binding protein hemopexin (Larsen et al. 2010; Dalko et al. 2015). These diseases frequently affect children with SAM in the developing world, and malaria in particular is significantly associated with malnutrition (Berkley et al. 2009; Khogali et al. 2011). Our findings indicate elevated heme during SAM may contribute to increased severity of these infections, and therefore treatments targeting free heme may be beneficial.
Our study has several limitations. Firstly, reliable information regarding other infections and pathologies was not available due to the limited local diagnostic facilities, so their possible impact on the microbiota and metabolome remains unknown. Children with malnutrition were also more likely to originate from rural settings, and therefore we cannot distinguish the role of environment from that of malnutrition with absolute certainty. However, we could not identify any metabolites that differed significantly between children from urban, peri-urban or rural settings, indicating residence does significantly impact the plasma metabolome in this cohort. Also, although we identified clear differences in the plasma metabolome of children according to nutritional status, this was not the only source of variation. We speculate that other unaccounted for sources of variation may include time of sample collection as it relates to circadian rhythms (Dallmann et al. 2012) and feeding time, diet and other environmental exposures, and/or the proximity of the plasma sampling to the plasma-red blood cell interface. The samples in this study were taken at random time points, with no requirements for fasting or feeding prior to collection. This may explain why we did not identify some of the classical signs of fasting, such as ketones elevated in the SAM group. However, our pragmatic collection method emphasized metabolites robust to external factors such as time of day and feeding. This strengthens the applicability of our findings to a clinical setting, where controlling for such variables may not be feasible.
In conclusion, we have demonstrated the plasma metabolome discriminates children with SAM from controls and identified a number of previously unreported metabolic derangements in malnutrition, providing new insight into disease mechanisms and management. Future studies are needed to monitor these metabolites during intervention to identify those most correlated with mortality and/or recovery. Validation of such biomarkers may enable better identification of children at highest risk of poor outcomes, and could guide improved management for a leading cause of childhood mortality worldwide.
References
Aitchison, J. (1982). The statistical analysis of compositional data. Journal of the Royal Statistical Society: Series B Statistical Methodology, 44, 139–177. doi:10.2307/2345821.
Allen, F., Pon, A., Wilson, M., et al. (2014). CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Research, 42, 94–99. doi:10.1093/nar/gku436.
Balla, G., Vercellotti, G. M., Muller-Eberhard, U., et al. (1991). Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Laboratory Investigation; A Journal of Technical Methods and Pathology, 64, 648–655.
Bartz, S., Mody, A., Hornik, C., et al. (2014). Severe acute malnutrition in childhood: Hormonal and metabolic status at presentation, response to treatment, and predictors of mortality. The Journal of Clinical Endocrinology and Metabolism, 99, 2128–2137. doi:10.1210/jc.2013-4018.
Benjamini, Y., Hochberg, Y. (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B Statistical Methodology 57, 289–300.
Berkley, J. A., Bejon, P., Mwangi, T., et al. (2009). HIV infection, malnutrition, and invasive bacterial infection among children with severe malaria. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America, 49, 336–343. doi:10.1086/600299.
Brown, E. M., Wlodarska, M., Willing, B. P., et al. (2015). Diet and specific microbial exposure trigger features of environmental enteropathy in a novel murine model. Nature Communications, 6, 7806. doi:10.1038/ncomms8806.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335–336. doi:10.1038/nmeth.f.303.
Caulfield, L. E., de Onis, M., Blössner, M., & Black, R. E. (2004). Undernutrition as an underlying cause of child deaths associated with diarrhea, pneumonia, malaria, and measles. The American Journal of Clinical Nutrition, 80, 193–198.
Cunnane, S. C., & Anderson, M. J. (1997). The majority of dietary linoleate in growing rats is beta-oxidized or stored in visceral fat. The Journal of Nutrition, 127, 146–152.
Dalko, E., Das, B., Herbert, F., et al. (2015). Multifaceted role of Heme during Severe Plasmodium falciparum infections in India. Infection and Immunity, 83, 3793–3799. doi:10.1128/IAI.00531-15.
Dallmann, R., Viola, A. U., Tarokh, L., et al. (2012). The human circadian metabolome. Proceedings of the National Academy of Sciences of the United States of America, 109, 2625–2629. doi:10.1073/pnas.1114410109.
Denno, D. M., VanBuskirk, K., Nelson, Z. C., et al. (2014). Use of the lactulose to mannitol ratio to evaluate childhood environmental enteric dysfunction: A systematic review. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America, 59(Suppl 4), S213–S219. doi:10.1093/cid/ciu541.
Dunn, W. B., Broadhurst, D., Begley, P., et al. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocols, 6, 1060–1083. doi:10.1038/nprot.2011.335.
Ebert, M. P., Niemeyer, D., Deininger, S. O., et al. (2006). Identification and confirmation of increased fibrinopeptide a serum protein levels in gastric cancer sera by magnet bead assisted MALDI-TOF mass spectrometry. Journal of Proteome Research, 5, 2152–2158. doi:10.1021/pr060011c.
Edwards, R. L., Levine, J. B., Green, R., et al. (1987). Activation of blood coagulation in Crohn’s disease. Increased plasma fibrinopeptide A levels and enhanced generation of monocyte tissue factor activity. Gastroenterology, 92, 329–337.
Eisenberg, P. R., Sherman L a., Schectman, K., et al. (1985). Fibrinopeptide A: A marker of acute coronary thrombosis. Circulation, 71, 912–918. doi:10.1161/01.CIR.71.5.912.
Fernandes, A. D., Macklaim, J. M., Linn, T. G., et al. (2013). ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-SEq. PLoS One, 8, e67019. doi:10.1371/journal.pone.0067019.
Fernandes, A. D., Reid, J. N., Macklaim, J. M., et al. (2014). Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16 S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome, 2, 15. doi:10.1186/2049-2618-2-15.
Gabbs, M., Leng, S., Devassy, J. G., et al. (2015). Advances in our understanding of oxylipins derived from dietary PUFAs. Advances in Nutrition, 6, 513–540. doi:10.3945/an.114.007732.
Ghisolfi, J., Charlet, P., Ser, N., et al. (1978). Plasma free amino acids in normal children and in patients with proteinocaloric malnutrition: Fasting and infection. Pediatric Research, 12, 912–917.
Gloor, G. B., Hummelen, R., Macklaim, J. M., et al. (2010). Microbiome profiling by illumina sequencing of combinatorial sequence-tagged PCR products. PLoS One, 5, e15406. doi:10.1371/journal.pone.0015406.
Hashimoto, T., Perlot, T., Rehman, A., et al. (2012). ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature, 487, 477–483. doi:10.1038/nature11228.
Holman, R. T., Johnson, S. B., Mercuri, O., et al. (1981). Essential fatty acid deficiency in malnourished children. The American Journal of Clinical Nutrition, 34, 1534–1539.
Jiang, P., Stanstrup, J., Thymann, T., et al. (2015). Progressive changes in the plasma metabolome during malnutrition in juvenile pigs. Journal of Proteome Research. doi:10.1021/acs.jproteome.5b00782.
Jones, K. D., Ali, R., Khasira, M., et al. (2015). Ready-to-use therapeutic food with elevated n-3 polyunsaturated fatty acid content, with or without fish oil, to treat severe acute malnutrition: A randomized controlled trial. BMC Medicine, 13, 93. doi:10.1186/s12916-015-0315-6.
Kar, B. R., Rao, S. L., & Chandramouli, B. A. (2008). Cognitive development in children with chronic protein energy malnutrition. Behavioral and Brain Functions, 4, 31.
Kessner, D., Chambers, M., Burke, R., et al. (2008). ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics (Oxford, England), 24, 2534–2536. doi:10.1093/bioinformatics/btn323.
Khogali, M., Zachariah, R., Keiluhu, A., et al. (2011). Detection of malaria in relation to fever and grade of malnutrition among malnourished children in Ethiopia. Public Health Action, 1, 16–18. doi:10.5588/pha.11.0008.
Koletzko, B., Abiodun, P. O., Laryea, M. D., & Bremer, H. J. (1986). Fatty acid composition of plasma lipids in Nigerian children with protein-energy malnutrition. European Journal of Pediatrics, 145, 109–115.
Krishna, S., Waller, D. W., ter Kuile, F., et al. (1994). Lactic acidosis and hypoglycaemia in children with severe malaria: Pathophysiological and prognostic significance. Transactions of the Royal Society of Tropical Medicine and Hygiene, 88, 67–73.
Kvissberg, M. A., Dalvi, P. S., Kerac, M., et al. (2016). Carbohydrate malabsorption in acutely malnourished children and infants: A systematic review. Nutrition Reviews, 74, 48–58. doi:10.1093/nutrit/nuv058.
Larsen, R., Gozzelino, R., Jeney, V., et al. (2010). A central role for free heme in the pathogenesis of severe sepsis. Science Translational Medicine, 2, 51ra71. doi:10.1126/scitranslmed.3001118.
Leichsenring, M., Sütterlin, N., Less, S., et al. (1995). Polyunsaturated fatty acids in erythrocyte and plasma lipids of children with severe protein-energy malnutrition. Acta Paediatrica (Oslo, Norway: 1992), 84, 516–520.
Medina, R., Wing, S. S., Haas, A., Goldberg, A. L. (1991). Activation of the ubiquitin-ATP-dependent proteolytic system in skeletal muscle during fasting and denervation atrophy. Biomedica Biochimic Acta, 50, 347–356.
Mikkelsen, M. E., Miltiades, A. N., Gaieski, D. F., et al. (2009). Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Critical Care Medicine, 37, 1670–1677. doi:10.1097/CCM.0b013e31819fcf68.
Mtove, G., Nadjm, B., Hendriksen, I. C. E., et al. (2011). Point-of-care measurement of blood lactate in children admitted with febrile illness to an African District Hospital. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America, 53, 548–554. doi:10.1093/cid/cir471.
Nagao, M., Parimoo, B., & Tanaka, K. (1993). Developmental, nutritional, and hormonal regulation of tissue-specific expression of the genes encoding various acyl-CoA dehydrogenases and alpha-subunit of electron transfer flavoprotein in rat. The Journal of Biological Chemistry, 268, 24114–24124.
Patti, G. J., Tautenhahn, R., & Siuzdak, G. (2012). Meta-analysis of untargeted metabolomic data from multiple profiling experiments. Nature Protocols, 7, 508–516. doi:10.1038/nprot.2011.454.
Pearson, A. D., Eastham, E. J., Laker, M. F., et al. (1982) Intestinal permeability in children with Crohn’s disease and coeliac disease. British Medical Journal (Clinical Research Edition), 285, 20–21. doi:10.1136/bmj.285.6334.20.
Reddy, A. J., Lam, S. W., Bauer, S. R., & Guzman, J. A. (2015). Lactic acidosis: Clinical implications and management strategies. Cleveland Clinic Journal of Medicine, 82, 615–624. doi:10.3949/ccjm.82a.14098.
Schaible, U. E., & Kaufmann, S. H. E. (2007). Malnutrition and Infection: Complex mechanisms and global impacts. PLoS Medicine, 4, e115. doi:10.1371/journal.pmed.0040115.
Seixas, E., Gozzelino, R., Chora, A., et al. (2009). Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proceedings of the National Academy of Sciences of the United States of America, 106, 15837–15842. doi:10.1073/pnas.0903419106.
Shapiro, N. I., Howell, M. D., Talmor, D., et al. (2005). Serum lactate as a predictor of mortality in emergency department patients with infection. Annals of Emergency Medicine, 45, 524–528. doi:10.1016/j.annemergmed.2004.12.006.
Smit, E. N., Muskiet, F. A. J., Boersma, E. R. (2004). The possible role of essential fatty acids in the pathophysiology of malnutrition: A review. Prostaglandins Leukot. Essential Fatty Acids, 71, 241–250.
Smith, C. A., O’Maille, G., Want, E. J., et al. (2005). METLIN: A metabolite mass spectral database. Therapeutic Drug Monitoring, 27, 747–751. doi:10.1097/01.ftd.0000179845.53213.39.
Smith L., & Gaya, D. R. (2012). Utility of faecal calprotectin analysis in adult inflammatory bowel disease. World Journal of Gastroenterology: WJG, 18, 6782–6789. doi:10.3748/wjg.v18.i46.6782.
Smith, M. I., Yatsunenko, T., Manary, M. J., et al. (2013). Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science, 339, 548–554. doi:10.1126/science.1229000.
Stein, S. E. (1999). An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. Journal of the American Society for Mass Spectrometry, 10, 770–781. doi:10.1016/S1044-0305(99)00047-1.
Styczynski, M. P., Moxley, J. F., Tong, L. V., et al. (2007). Systematic identification of conserved metabolites in GC/MS data for metabolomics and biomarker discovery. Analytical Chemistry, 79, 966–973. doi:10.1021/ac0614846.
Subramanian, S., Huq, S., Yatsunenko, T., et al. (2014). Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature, 509, 417–421. doi:10.1038/nature13421.
Sutherland, L. R., Verhoef, M., Wallace, J. L., et al. (1994). A simple, non-invasive marker of gastric damage: Sucrose permeability. Lancet, 343, 998–1000. doi:10.1016/S0140-6736(94)90125-2.
Timotej, V. (2011) A comparison of parameters below the limit of detection in geochemical analyses by substitution methods. RMZ Mater Geoenvironment, 58, 393–404.
Wagener, F. A., Eggert, A., Boerman, O. C., et al. (2001). Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood, 98, 1802–1811. doi:10.1182/blood.V98.6.1802.
WHO. (2009). WHO child growth standards and the identification of severe acute malnutrition in infants and children. WHO Libr 1–12.
Wishart, D. S., Tzur, D., Knox, C., et al. (2007). HMDB: The Human Metabolome Database. Nucleic Acids Research, 35, D521–D526. doi:10.1093/nar/gkl923.
Zhang, M. H., Xu, X. H., Wang, Y., et al. (2013). A prognostic biomarker for gastric cancer with lymph node metastases. The Anatomical Record Advances in Integrative Anatomy and Evolutionary Biology, 296, 590–594. doi:10.1002/Ar.22642.
Acknowledgements
The authors appreciate the support of Dr. Folake Samuel of the Department of Human Nutrition, University of Ibadan, UNICEF for supply of RUTF to the patients and Dr. A. A. Oloko of the Federal Medical Centre, Gusau for assistance during data collection and management of the patients. We are grateful to Linda Thomas, Yakult Ltd UK, for providing financial support for the project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests in relation to the work described.
Ethical approval
This study was approved by the Joint Ethical Review Committees of the University of Ibadan/University College Hospital, Ibadan, Nigeria, and was conducted in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Human stool and peripheral blood samples were collected after informed consent from all parents of children enrolled in the study.
Additional information
Amy McMillan and Adebola E. Orimadegun are joint first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McMillan, A., Orimadegun, A.E., Sumarah, M.W. et al. Metabolic derangements identified through untargeted metabolomics in a cross-sectional study of Nigerian children with severe acute malnutrition. Metabolomics 13, 13 (2017). https://doi.org/10.1007/s11306-016-1150-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-016-1150-2