
Abstract
Over the last seven years our laboratory has focused on the determination of the structural aspects of nucleoside triphosphate diphosphohydrolases (NTPDases) using site-directed mutagenesis and computational comparative protein modeling to generate hypotheses and models for the hydrolytic site and enzymatic mechanism of the family of NTPDase nucleotidases. This review summarizes these studies utilizing NTPDase3 (also known as CD39L3 and HB6), an NTPDase family member that is intermediate in its characteristics between the more widely distributed and studied NTPDase1 (also known as CD39) and NTPDase2 (also known as CD39L1 and ecto-ATPase) enzymes. Relevant site-directed mutagenesis studies of other NTPDases are also discussed and compared to NTPDase3 results. It is anticipated that many of the results and conclusions reached via studies of NTPDase3 will be relevant to understanding the structure and enzymatic mechanism of all the cell-surface members of this family (NTPDase1–3, 8), and that understanding these NTPDase enzymes will aid in modulating the many varied processes under purinergic signaling control. This review also integrates the site-directed mutagenesis results with a recent 3-D structural model for the extracellular portion of NTPDases that helps explain the importance of the apyrase conserved regions (ACRs) of the NTPDases. Utilizing this model and published work from Dr Guidotti’s laboratory concerning the importance and characteristics of the two transmembrane helices and their movements in response to substrate, we present a speculative cartoon model of the enzymatic mechanism of the membrane-bound NTPDases that integrates movements of the extracellular region required for catalysis with movements of the N- and C-terminal transmembrane helices that are important for control and modulation of enzyme activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Site-directed mutagenesis studies (point mutations) of NTPDases
In this section site-directed mutagenesis studies introducing point mutations in NTPDases are reviewed. Other manipulations of protein structure including chimeras and truncation mutants of NTPDases are not covered in this review.
NTPDase3 studies (see Table 1)
Over the last seven years, 10 studies have been published using point mutations introduced by site-directed mutagenesis to investigate the structure and function of human NTPDase3 (see Table 1). What follows is a concise summary of those studies, in chronological order.
The first report of site-directed mutagenesis of any NTPDase [1] was important for establishing experimentally what had been hypothesized based on limited sequence homology of the NTPDases with the actin/heat shock protein/sugar kinase superfamily of proteins. Specifically, the conservation of the two ‘DXG’ phosphate binding motifs of the actin superfamily in the NTPDases suggested that these regions serve the same function in the NTPDases as they do in the actin family. The importance of these residues in ACR1 and ACR4 in the NTPDases was established by mutation of these aspartic acids and glycines, resulting in the inactivation of the enzyme without a change in the protein expression level.
The significance of the two tryptophan residues that are most highly conserved in the NTPDases was established through the mutation of W187 and W459 in NTPDase3 [2]. Mutation of W187 in ACR3 resulted in inactivation, while mutation of W459 in ACR5 resulted in an active enzyme with increased ATPase activity, and therefore an increased ATPase:ADPase hydrolysis ratio. Coupling this W459A mutation with the D219E mutation in DXG2/ACR4 resulted in near conversion of NTPDase3 from an apyrase to an ecto-ATPase (i.e., increased dramatically the NTPase: NDPase hydrolysis ratio for most nucleotides), indicating that a major change in nucleotide tri-versus di-phosphatase specificity is possible with only a few changes in amino acid sequence.
Mutation of several conserved amino acids located in ACR1–4 established the importance of these regions and some of these residues for enzymatic activity of NTPDase3 [3]. The N191A mutation, located in ACR3, changed the ATPase:ADPase ratio to a similar value as exhibited by NTPDase1 (approximately 1.0). Mutation of a serine residue in ACR4 (S224A) resulted in complete inactivation of enzyme activity. Mutation of a glutamic acid in ACR3 (E182) to either aspartic acid or glutamine also caused complete inactivation of activity. However, differences were noted in the two mutations at this site, with the E182D mutant incapable of binding to the nucleotide analogue, Cibacron blue, as well as being more susceptible to limited proteolysis, while the E182Q mutant behaved more like wild-type NTPDase3 in these assays. These results, along with a lower expression level for the E182D mutant, suggest that this residue in ACR3 may be important both for hydrolysis of substrate and for proper folding of the protein. This is consistent with the location of ACR3 in the 3-D computational model of the extracellular portion of NTPDase3 shown in Figure 1B and Figure 2.

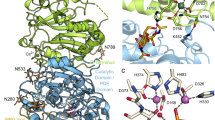
Models of NTPDase3. Panel A — Cartoon model of NTPDase3, showing the five extracellular disulfide bonds which are presumably conserved in all cell surface NTPDases (NTPDase1–3, 8), as well as the ACR regions and the free sulfhydryls existing in the TM helices and the N-terminal cytoplasmic tail. Panel B — FUGUE generated 3-D model of the extracellular portion of NTPDase3 based on the PPX/GPPA protein template structure. The location of the ACR regions and the cysteine residues included in the model are indicated. Panel C — LOOPP 3-D model of the internal NTPDase3 domain. The sequence between ACR4 and ACR5 (approximately residues 240–380, encircled in purple in Figure 1A) was used to identify a tyrosine phosphatase having some fold similarity to this region of NTPDase3. The resultant 3-D model is of interest since it places all six cysteine residues in this part of the NTPDase3 sequence in positions consistent with the determined second, third, and fourth disulfide bonds. (Figure 1A, B were modified from Figure 8, and Figure 1C was modified from Supporting Information file #6 named “LOOPP_1P8A_NTPDase3_domain_alignment_and_model.jpg”, all appearing in [18], and reproduced with permission from Biochemistry 2005, 44, 8998–9012, copyright 2005, Am. Chem. Soc.).

NTPDase3 inactivating mutations and ACR regions superimposed on the 3-D model. The FUGUE generated homology model for human NTPDase3, based on the structure of the PPX/GPPA bacterial enzyme template, PDB code 1T6C, is shown [18]. Most of the protein is shown in wireframe representation (thin gray lines), while the ACR regions are shown as colored ribbon structures, and many of the inactivating mutations of NTPDase3 listed in Table 1 are represented in red space filling mode (all are in ACRs). The 2 ‘DXG’ phosphate binding motifs contained within the blue ACR1 (D62 and G64) and ACR4 (D219 and G221) beta sheet structures define the right and left sides of the enzymatic cleft, accessed by substrate from the upper part of the structure. Also in the active site are S224 (in the blue ACR4 beta sheet) and E182 (in the green ACR3 alpha helix). A conserved tryptophan (W187) also resides in the green ACR3 helix, which along with the yellow ACR5 alpha helix, connect the two lobes of the extracellular NTPDase3 structure. Also indicated in the figure are the ACR1a and ACR4a regions (purple). ACR4a contains both C261 and G263 residues. C261 is part of the second of five conserved NTPDase extracellular disulfide bonds [18]. G263 was found to be critical for proper folding and processing of NTPDase3 [8]. The boundaries of the ACR regions are defined by the peaks in the PlotSimilarity multiple sequence analysis (see Figure 7 in [8]). For human NTPDase3, the residues comprising the ACR regions are: ACR1, K56–K79; ACR1a, V94–Y102; ACR2, T132-L145; ACR3, I176-G195; ACR4, G216-F229; ACR4a, Y255-A267; and ACR5, W459-P472.
Several conserved histidine residues were mutated in NTPDase3 [4]. This study was initiated since it had been previously noted that a chemical modification reagent that is selective for histidine, diethylpyrocarbonate (DEPC), was one of the few amino acid selective modification reagents that inhibited the NTPDases [5, 6]. Surprisingly, no conserved histidine residue was found to be essential for activity or to be the site of DEPC induced inhibition. However, H135 was found to be important for full activity, and mutagenesis of H257 resulted in greater sensitivity to DEPC inactivation. The R67H mutation, which makes NTPDase3 more like the other NTPDases, which have a histidine in this position in ACR1, increased the sensitivity to inactivation by DEPC. This implies that, in other NTPDases, this histidine in ACR1 may be a major site of modification by DEPC causing inactivation, as was suggested by Dzhandzhugazyan and Plesner [7] for NTPDase1/CD39, based on its conservation in NTPDases other than NTPDase3. Interestingly, simultaneous replacement of all three of the most highly conserved histidine residues in NTPDase3 caused the formation of large aggregates, without inactivating the enzyme, suggesting that these histidines may be important for higher order oligomeric formation or stability.
The most highly conserved glycine residues (excluding the 2 DXG glycines mutated in NTPDase3 discussed above [1]) were mutated to alanine [8]. One rationale for making these mutations was the observation that several conserved glycines were important for the structure and function of the actin superfamily of proteins [9, 10]. Indeed, several such residues were found to be important for NTPDase3 activity (see Table 1). In addition, two of the mutants, G263A and G462A, resulted in moderate to severe problems in folding and/or processing of NTPDase3. This study was also significant for the identification of two additional ACR regions (each containing a conserved glycine residue), termed ACR1a and ACR4a. These regions were identified by objective and quantitative computer analyses of the multiple sequence alignment for all the NTPDase sequences available at that time.
The significance of the three non-extracellular cysteine residues, assumed to exist as free sulfhydryls in NTPDase3, were investigated by mutation to serine [11]. C10, located in the N-terminal cytoplasmic tail region, was found to be responsible for the covalent dimer formation that occurs by spontaneous oxidation of that residue during membrane preparation. This C10S mutant would be used in several subsequent studies as a replacement for the wild-type NTPDase3, since the C10S mutant has wild-type like activity and characteristics, but does not spontaneously form covalent dimers, which obscures the interpretation of some experiments. C501, located in the C-terminal membrane spanning (MS) helix of NTPDase3, was found to be the site of labeling by p-chloromercuriphenylsulfonic acid (pCMPS) that results in partial enzyme inactivation. This was somewhat unexpected, since C501 is predicted to be located almost in the middle of the C-terminal transmembrane (TM) domain, and should therefore be inaccessible to this charged reagent. This suggested the possibility of an aqueous pore or crevice, allowing pCMPS to reach C501. This finding is consistent with more recent results published by Grinthal and Guidotti [12], suggesting that the extracellular portions of the TM helices are more closely associated with one another and have less flexibility than the cytoplasmic portions of the same transmembrane helices (see below). Thus, it seems likely that the pCMPS reagent accesses C501 via the cytoplasmic face of the membrane rather than the extracellular face of the membrane (see Figure 3).

Model of the speculative essential connection between movements in the transmembrane helices and the N- and C-terminal extracellular lobes seen in Figure 1B. Transmembrane helices are represented by cylinders, the cell membrane as a yellow box, and the N- and C-terminal cytoplasmic tails are indicated. The extracellular region is from the FUGUE model shown in Figure 1B. The extracellular N-terminal lobe is green, and the extracellular C-terminal lobe is cyan in color. The DXG motifs in ACR1 and ACR4 beta sheets are in dark blue space filling mode. The ACR1 beta sheet and the ACR5 alpha helix, which connect the extracellular lobes to their respective transmembrane helices, are colored red. The conserved proline residues near the interfaces between the TM helices and ACR1 and ACR5 regions are represented by ‘P’. This model speculates that the movement of the N- and C-terminal lobes of the extracellular portion of NTPDase3 (seen in Figure 1B) is structurally and functionally linked to the movements of the N- and C-terminal TM helices, as first described by Grinthal and Guidotti [12]. These coordinated movements result in, from left to right, the availability of substrate to the active site cleft, the movement of the ACR1 and ACR4 regions into close contact with the substrate to allow catalysis (the movement of the cyan colored C-terminal lobe is mediated by movement of the ACR5 alpha helix, which is in turn, is linked to the movement of the C-terminal membrane spanning helix via the intervening conserved proline residues), and the re-opening of the cleft to allow the products to be released and a new cycle of hydrolysis to begin. C501, the site of modification by pCMPS resulting in partial inactivation of NTPDase3 [11], is depicted to become accessible to this reagent by movement of the TM helices, which is coupled to the movement of the extracellular lobes that is required for enzymatic activity. The other monomer of NTPDase3 involved in the catalytic dimer is not shown for the sake of clarity. The oligomeric nature of the cell surface NTPDases will need to incorporated in more advanced models generated as a result of future studies.
The role of glycosylation at the most conserved glycosylation site of the NTPDases, which is located between ACR1 and ACR1a, was established by mutation of N81 and T83 in NTPDase3 [13]. Mutation of either the asparagine residue where the sugars are attached (N81D and N81E) or the threonine residue that is part of the consensus sequence for glycosylation at that site (T83A) resulted in decreased nucleotidase activities. The ATPase activity was more severely affected than the ADPase activity, and the Mg2+ activity was more severely decreased than the Ca2+ activity, suggesting that the mutations might be decreasing activity via oligomeric structure effects. Loss of inhibition by a deglycosylation enzyme (PNGase-F) and the lack of stimulation/stabilization of activity by the lectin, Concanavalin A, were also observed in these mutants.
The importance of lysine residue 79 located near the conserved glycosylation site near ACR1 was investigated [14]. This residue is conserved in cell surface NTPDases (NTPDase1–3, 8), but not in the soluble (NTPDase5–6) and intracellular (NTPDase4, 7) members of this nucleotidase family (see Table 1 in [14]). All substitutions at this site resulted in a loss of 70%–80% of activity. Only K79R, which alone maintains the positive charge at this residue, remained able to be stimulated/stabilized by the lectin Con A (presumably via binding at the nearby N81 glycosylation site [13]). The residual activity of all the mutants at the K79 site became resistant to the inhibitory effects of the detergent, Triton X-100. Since this detergent was shown to dissociate the NTPDases into monomers (presumably its mechanism of inhibition of activity [11, 15]), it was postulated that these K79 mutants might have weakened oligomeric structures, accounting for their lower activity and insensitivity to Triton X-100. It was also hypothesized that the reason for the activity of these mutants being higher in Ca2+ than in Mg2+ might be due to a subtle stabilization of tertiary or quaternary structure by Ca2+, partially correcting the destabilization of these mutants caused by substitution at K79.
The residues responsible for the increase in activity observed upon acetylation of NTPDase3 with the tyrosine selective chemical modification reagent, N-acetylimidazole (N-AI) were established by site-directed mutagenesis [16]. N-AI modification resulted in a larger increase in ATPase (approximately 2-fold) than ADPase (approximately 1.4 fold) activity in wild-type NTPDase3. However, this reagent did not increase the activity of other NTPDases tested. In NTPDase3, mutation of Y252 (in ACR4a) did not negatively affect activity, but abolished the N-AI induced increase in ATPase and ADPase activity. This effect of N-AI was also abolished by prior solubilization with the monomerizing detergent Triton X-100, and reversed by subsequent treatment with Triton X-100, suggesting that the effect of N-AI modification is dependent upon changes in the tertiary and/or quaternary structure which result in a more stable oligomeric structure with more stable enzyme activity. In addition, the mutations that abolished the N-AI induced increase in nucleotidase activity also diminished the increase in activity observed after treatment with Con A or a chemical cross-linker. These observations, coupled with the nearby location of a computer predicted [17] helix-loop-helix dimerization domain (238NTSDOMQVS246) lead to the hypothesis that this region near ACR4a may be important for the oligomeric structure and stability of NTPDase3.
Site directed mutagenesis of the 10 conserved extracellular cysteines (which presumably form five conserved disulfide bonds in all the cell-surface NTPDases) established the location of the five extracellular disulfide bonds in NTPDase3 ([18], see Figure 1A). Unlike soluble NTPDase6, whose two disulfide bonds were determined by classic protein chemistry techniques [19], the membranous NTPDases are not amenable to this direct approach. Therefore, the disulfides were assigned by analysis of the enzymatic phenotypes of all the single and double extracellular cysteine mutants of NTPDase3 [18]. Thus, the expressed cysteine mutant proteins were analyzed for nucleotidase activity, glycosylation state and sensitivity to Endo H, delivery to the cell surface, and susceptibility to inactivation by reduction, in order to deduce the disulfide bond architecture. Loss of each of the five disulfide bonds had distinct effects on enzyme activity, processing, and structure (see Table 1 and [18]).
The same paper that examined the disulfide bond structure of NTPDase3 also generated three-dimensional models of NTPDase3 (and other NTPDases) via homology modeling of the NTPDase3 extracellular sequence [18]. A high degree of structural fold similarity between the NTPDases and a bacterial exopolyphosphatase (PDB 1T6C) was observed [18]. The resultant theoretical 3-D models of the extracellular portion of NTPDase3 (see [18] and the discussion below), based on homology with this exopolyphosphatase, were consistent with the assignment of the disulfide bonds occurring in regions of good fold similarity between NTPDase3 and the exopolyphosphatase. The 3-D models also suggested the structural basis for the importance of several apyrase conserved regions of the NTPDases (see Figure 1B).
Relevant mutagenesis studies using other NTPDases (see Table 2)
Several site-directed mutagenesis studies (as well as other manipulations of the protein structure) using NTPDase1 (CD39) have been published by Dr Guidotti’s group. One early example is the work demonstrating the importance of a histidine residue (H59) in ACR1 of rat brain NTPDase1 [20] (this residue is R67 in rat, mouse, and human NTPDase3). The more recent studies from the Guidotti laboratory have focused on the importance of the presence and motility of the two transmembrane helices in rat brain NTPDase1/CD39 that are common to all the cell-surface NTPDases [12, 15, 20, 21], as well as the significance of glycosylation for NTPDase1 processing and function [22, 23]. Since Dr Guidotti is also contributing an article to this special issue on nucleotidases, most of his laboratory’s work in this field will not be discussed here. However, one recent study from his laboratory [12] is of particular relevance to this review. In that study, the authors introduced cysteines into the transmembrane (TM) domains of rat brain CD39/NTPDase1 and used a disulfide cross-linking strategy to investigate TM helix interactions and dynamics (and their functional consequences). They concluded that strong interactions exist between the regions of the TM domains nearest the extracellular side of the membrane, while only weak interactions exist near the cytoplasmic side of the TM domains. They also presented evidence that NTPDase1 activity is dependent on the ability of these two TM helices to move relative to one another.
Dr Marcus’s group has also published mutagenesis studies of CD39/NTPDase1. They mutagenized conserved amino acids in ACR1-4 in a soluble construct derived from human CD39/NTPDase1 [24]. Some of the mutations made in that study are homologous to the residues mutated in NTPDase3 (see Tables 1 and 2). They concluded that NTPDase1 E174 (equivalent to E182 in NTPDase3) and S218 (equivalent to S224 in NTPDase3) are essential for the enzymatic and biological (inhibition of ADP-induced platelet aggregation) activities of solCD39/NTPDase1. They also noted that the S57A mutation in ACR1 caused a two-fold increase in ADPase activity, but no change in ATPase activity, and that the Y127A mutation in ACR2 lost about 50% ADPase and ATPase activities. In addition, differences in preference for Ca2+ versus Mg2+ were noted for some mutants.
The aspartic acid residues constituting the ‘D’ residues in both ‘DXG’ motifs in a soluble form of human CD39/NTPDase1 (D54 and D213, corresponding to D62 and D219 in NTPDase3) were mutated by Drosopoulos [25]. In contrast to our laboratory’s results using NTPDase3 (see [1], Table 1), mutation of these residues did not cause nearly total inactivation of NTPDase1 ATPase and ADPase activities, using either Ca2+ or Mg2+ as co-substrate. Only when both residues were mutated was the activity completely abolished. Kinetic analyses of the single mutants revealed changes in Km for Ca2+ and nucleotides. It was concluded that these two residues in the DXG regions are involved in Ca2+ utilization, and possibly involved in coordination of the Ca2+ used as co-substrate with the nucleotide. Our laboratory has re-examined these homologous mutations in NTPDase3 trying to mimic assay conditions used by Drosopoulos, and we have obtained basically the same results as were previously published [1] — in NTPDase3, these mutants are basically inactive using either calcium or magnesium as co-substrate, using a variety of nucleotide concentrations and assay buffer compositions. Thus, it is possible that there may be some real differences in the roles of the DXG motifs between the membrane-bound NTPDase3 and the soluble NTPDase1 used in these two seemingly partially contradictory studies. Interestingly, we note that the amino acid residues homologous to NTPDase ACR4 form a Ca2+ binding site in the crystal structure of the bacterial exopolyphosphatase (PPX/GPPA [26]) used as a template for construction of the computational model of the 3-D structure of the NTPDases (see below).
Mateo et al. [27] deduced the importance of C399 and N443 in human NTPDase2/ecto-ATPase while studying splice variants of this ecto-ATPase. The C399S mutant resulted in a totally inactive protein, while the N443D mutant, which removes a potential glycosylation site, had reduced activity. Those authors concluded that both these mutations reduced enzyme activity via their detrimental effects on the folding and processing of these proteins, presumably explaining why, of the three splice mutants of human NTPDase2 examined, only the longest protein (495 amino acids) has any nucleotidase activity. They further deduced that the mutation of C399 most likely resulted in an inactive enzyme due to disruption of a disulfide bond necessary for maintaining the structure of the enzyme. This result and conclusion is consistent with our laboratory’s findings that the cysteine residue corresponding to C399 in NTPDase2 is indeed disulfide bonded, both in soluble NTPDase6 (equivalent to C354 in NTPDase6, in the disulfide bond C340–C354 [19]), and in NTPDase3 (equivalent to C422 in NTPDase3, in the 5th disulfide bond C399–C422 [18]), and that this disulfide bond is essential for processing and activity in NTPDase3 [18].
Structural modeling of the NTPDases
The location of the two disulfide bonds in the soluble NTPDase6 were determined by direct protein chemistry techniques [19]. Unfortunately, this direct approach is not feasible for the cell-surface localized NTPDase3, due to the higher complexity (5 versus 2 disulfide bonds) and lack of sufficient amounts of purified protein. Thus, we were forced to establish likely disulfide bonds using an indirect mutagenesis approach [18]. Even though the disulfide bonds were determined indirectly, the disulfide bond assignments made are likely to be correct due to the existence of several types of data that are all consistent with one another and with the assignments made [18], and they are likely to be present in all the cell surface NTPDases, since these 10 extracellular cysteines are conserved. The disulfide bond assignments are schematically indicated in Figure 1A. We then used this information to help evaluate a computational model of the 3-D structure of NTPDase3, which was obtained by threading the amino acid sequences of the extracellular portions of several NTPDases through a protein database, using fold recognition software (the FUGUE and FFAS programs, [18]). Only one protein was found to have very good fold similarity to the NTPDases, a bacterial exopolyphosphatase (PPX/GPPA). A two dimensional representation of the NTPDase3 3-D model based on the PPX/GPPA template structure is shown in Figure 1B.
As objectively evaluated by the program Verify3D “http://www.doe-mbi.ucla.edu/Services/Verify_3D/ [28]), some parts of the resulting 3-D FUGUE “http://wwwcryst. bioc.cam.ac.uk/fugue/ [29]) NTPDase3 model are more likely to be valid than others. Thus, the N-terminal lobe of the molecule, containing ACR1, ACR1a, and ACR2, as well as the C92–C116 disulfide bond, is likely to be close to the real structure of NTPDase3. In addition, ACR3, ACR4 and the region immediately preceding ACR5 are also predicted to be well modeled by the Verify3D program (see Supporting Information file # 3 in [18]). However, there is a region of the protein after ACR4 and before ACR5 where the template protein (bacterial exopolyphosphatase (PPX/GPPA)) differs in predicted secondary structure and predicted folding from the NTPDases. This region includes C261 and C308, which are disulfide linked, but are not near each other in the model (see Figure 1B). This is in contrast to C92/C116 and C399/C422, which are close together in the model, as would be expected, since they are disulfide linked. Note that four cysteine residues in NTPDase3 (C289, C334, C347, and C353) are not represented in this model, since they have no corresponding residues in the PPX/GPPA template protein and therefore are not included in the model shown in Figure 1B (see [18]).
Fold recognition searches using the sequence of human NTPDase3 between ACR4 and ACR5 (from approximately residues 240–380, including the four cysteine residues not represented in the above model, which sequence was computationally predicted to form an independent domain [18]) as a query sequence revealed a number of weak fold similarity matches to a tyrosine phosphatase and other structurally related proteins. Although this domain’s fold similarity match was significantly worse than that found for the entire NTPDase3 molecule and the PPX/GPPA protein, a LOOPP (http://cbsu.tc.cornell.edu/software/loopp/ [30]) generated 3-D model of this putative domain of NTPDase3 based on a bacterial tyrosine phosphatase template (PDB code 1P8A) places the six NTPDase3 cysteines that exist in this part of NTPDase3 in positions consistent with the second (C261–C308), third (C289–C334), and fourth (C347–C355) disulfide bonds determined for NTPDase3 (see Figure 1C). Thus, the identified disulfide structure aids in the evaluation of the generated 3-D models of NTPDase3, and will be instrumental in further refinements and validation of these and other models.
The NTPDase3 model suggests the structural basis for the importance of several ACRs (see Figure 1B). ACR1 and ACR4 are each represented by a β-hairpin loop located on either side of a large cleft forming the active site (the highly conserved DXG sequences within ACR1 and ACR4 are depicted in blue space filling mode). ACR3 and ACR5 precisely correspond to two a-helices that connect the N- and C-domains (‘lobes’). Such a structure would allow substantial rotational movements of these two extracellular domains/lobes relative to one another. This motion has been described for the enzymes of the actin superfamily as a ‘butterfly-like’ opening of the active site cleft involved in catalysis [31, 32].
The NTPDase3 computational model is also consistent with the site-directed mutagenesis studies listed in Tables 1 and 2. In Figure 2, we highlight the mutations of NTPDase3 that cause nearly complete inactivation of nucleotidase activity (represented in red space-filling mode), in the context of the computational 3-D model of the extracellular portion of NTPDase3 and the ACR regions (represented as color-coded ribbons). The location of most of these inactivating mutations are clustered around the putative active site cleft formed by the two lobes of the theoretical structure. The two putative phosphate binding domains (DXG sequences), which are part of ACR1 and ACR4 beta sheet structures (in blue) are located on either side of the active site cleft. ACR3 (green ribbon) and ACR5 (yellow ribbon) each contain ‘an essential’ residue identified by mutagenesis, and constitute the alpha helices which connect the N-terminal and C-terminal lobes of the extracellular domain. ACR2 is shown in orange (behind ACR1 in Figure 2), and is part b-sheet and part a-helix. Two other ACR regions identified by quantitative multiple sequence alignments (ACR1a and ACR4a [8], in purple) map to the top of the two lobes of the structure, removed from the active site. ACR4a contains G263, mutation of which was found to completely inactivate the enzyme, apparently via its negative effects on protein folding and/or processing [8]. This region also contains C261, part of the second conserved disulfide bond in NTPDase3 [18]. Elimination of this bond by mutation of this residue not only causes substantial inactivation of activity, but also makes the small amount of residual activity very sensitive to DTT-induced inactivation. This suggests that this bond and region are important for the integrity of this part of the structure, and that absence of this disulfide bond opens up the C-terminal lobe to reduction of other disulfides in the structure, whose reduction by DTT causes inactivation of the residual NTPDase3 activity [18].
Thus, the computational model and the mutagenesis results are consistent with one another and both support a functional homology of the NTPDases with the actin superfamily of proteins. Therefore, it is logical to assume that the ‘butterfly-like’ opening of the active site cleft involved in catalysis for the actin superfamily [31, 32] also exists in the NTPDases. In concordance with Grinthal and Guidotti [12], we also speculate that these putative movements of the two extracellular domain lobes in the integral membrane NTPDases are functionally coupled to movements of the N- and C-terminal membrane spanning helices described by those authors. Furthermore, we formalize and expand upon the textual model of Grinthal and Guidotti [12] by constructing the cartoon model presented in Figure 3, which incorporates several additional features not described previously [12]. First, we incorporate our computational model of the 3-D structure of the extracellular region of the NTPDases (see Figures 1B and 2). Second, we note that there are conserved proline residues strategically located near the boundaries of the extracellular domain and the cell membrane, between the N-terminal TM helix and the ACR1 β-sheet, as well as between the C-terminal TM helix and the ACR5 α-helix (ACR1 and ACR5 are shown in red in Figure 3). We speculate that these prolines (represented by ‘P’ in Figure 3) may be important for providing the proper conformation and rigidity of structure needed for linking movements of the extracellular lobes (via ACR1 and ACR5) to movements of the transmembrane helices, and vice versa. These coordinated movements mediated through these conserved proline residues are speculated to be essential for full enzymatic activity. Thus, our cartoon model envisions the movement of the N- and C-terminal TM helices as being structurally and functionally linked to the movement of the N- and C-terminal lobes of the computational 3-D structure of the extracellular portion of NTPDase3, in agreement with an earlier report [12]. Third, this speculative cartoon model of NTPDase3 (Figure 3) is consistent with our previous study showing that C501, located in the Cterminal transmembrane domain, is accessible to the membrane impermeant reagent pCMPS, and that this modification reduces NTPDase3 activity [11]. The linked, coordinated movements of the TM helices and the extracellular domains permit the ACR1 and ACR4 regions to move into close contact with the substrate to allow catalysis (middle image in Figure 3), and then reopen to permit the release of product and a new cycle of hydrolysis to begin. Since these functionally important movements of the TM helices described for CD39/NTPDase1 [12] are very likely to occur in all the cell surface NTPDases (NTPDase1–3 and 8), and computational modeling of all the extracellular regions of the NTPDases (NTPDase1–3, 5–6, 8) gave rise to very similar predicted structures to that structure shown in Figure 1B for NTPDase3 (see Supporting Information file named NTPDase_FUGUE_Models.jpg in reference [18]), we propose that the speculative model depicted in Figure 3 is applicable to all the cell surface localized members of the NTPDase family.
Future directions for structural determination of the NTPDases
There are several future approaches for advancing our knowledge of the structure of the NTPDases and how their structures determine their functions. First, there is a need for refinement and experimental verification of the PPX/GPPA derived NTPDase 3-D computational models. Second, such a refined model based on the PPX/GPPA protein can then be combined with a second model which better approximates the structure of the region between ACR4 and ACR5 (based on a protein tyrosine phosphatase?) to generate a more accurate 3-D model of the entire extracellular region. Third, this combined model can then be used to design experiments for the purpose of elucidating the contact surfaces of the extracellular regions between NTPDase monomers that are important for oligomer formation and stability, and therefore for function. Fourth, the possibility that the NTPDase ACR4 region functions as a Ca2+ binding site, which is suggested by the PPX/GPPA crystal structure and by the human solCD39 D213 mutagenesis results [25], should be experimentally addressed. Fifth, crystallization and X-ray diffraction analysis and/or NMR structural determination of the naturally soluble form of NTPDase6 or some construct consisting of the extracellular region of another NTPDase can be used to experimentally validate any models developed, and will aid in the interpretation of all mutagenesis studies. Finally, the speculative role of the conserved proline residues near the boundaries between the N- and C-terminal transmembrane helices and the extracellular domain (see Figure 3) should be experimentally addressed as part of a larger effort to elucidate the role played by the proposed interconnected movements of the extracellular and transmembrane domains for the enzymatic mechanism and modulation of the oligomeric, integral membrane protein NTPDases.
Abbreviations
- ACR:
-
apyrase conserved region
- Con A:
-
Concanavalin A
- DTT:
-
dithiothreitol
- Endo H:
-
endoglycosidase H
- NTPDase3:
-
nucleotide triphosphate diphosphohydrolase 3
- pCMPS:
-
p-chloromercuriphenylsulfonic acid
- PDB:
-
protein data base
- PNGase-F:
-
peptide N-glycosidase F
- PPX/GPPA:
-
exopolyphosphatase/guanosine pentaphosphate phosphohydrolase
- TM:
-
transmembrane
References
Smith TM, Kirley TL. Site-directed mutagenesis of a human brain ecto-apyrase: Evidence that the E-type ATPases are related to the Actin/ Heat Shock 70/ Sugar Kinase Superfamily. Biochemistry 1999; 38: 321–28.
Smith TM, Lewis Carl SA, Kirley TL. Mutagenesis of two conserved tryptophan residues of the E-type ATPases: Inactivation and conversion of an ecto-apyrase to an ecto-NTPase. Biochemistry 1999; 38: 5849–57.
Yang F, Hicks-Berger CA, Smith TM et al. Site-directed mutagenesis of human nucleoside triphosphate diphosphohydrolase 3: The importance of residues in the apyrase conserved regions. Biochemistry 2001; 40: 3943–50.
Hicks-Berger CA, Yang F, Smith TM et al. The importance of histidine residues in human ecto-nucleoside triphosphate diphoshohydrolase-3 as determined by site-directed mutagenesis. Biochim Biophys Acta 2001; 1547: 72–81.
Kirley TL. Purification and characterization of the Mg2+-ATPase from rabbit skeletal muscle transverse tubule. J Biol Chem 1988; 263: 12682–89.
Saborido A, Moro G, Megias A. Transverse tubule Mg2+-ATPase of skeletal muscle — Evidence for extracellular orientation of the chicken and rabbit enzymes. J Biol Chem 1991; 266: 23490–98.
Dzhandzhugazyan KN, Plesner L. Diethyl pyrocarbonate inactivates CD39/ecto-ATPDase by modifying his-59. Biochim Biophys Acta 2000; 1466: 267–77.
Kirley TL, Yang F, Ivanenkov VV. Site-directed mutagenesis of human nucleoside triphosphate diphosphohydrolase 3: The importance of conserved glycine residues and the identification of additional conserved protein motifs in eNTPDases. Arch Biochem Biophys 2001; 395: 94–102.
Zeng C, Aleshin AE, Hardie JB et al. ATP-binding site of human brain hexokinase as studied by molecular modeling and site-directed mutagenesis. Biochemistry 1996; 35: 13157–64.
Zeng C, Aleshin AE, Chen G et al. The roles of glycine residues in the ATP binding site of human brain hexokinase. J Biol Chem 1998; 273: 700–4.
Murphy DM, Ivanenkov VV, Kirley TL. Identification of cysteine residues responsible for oxidative cross-linking and chemical inhibition of human nucleoside triphosphate diphosphohydrolase 3. J Biol Chem 2002; 277: 6162–69.
Grinthal A, Guidotti G. Dynamic motions of CD39 transmembrane domains regulate and are regulated by the enzymatic active site. Biochemistry 2004; 43: 13849–58.
Murphy DM, Kirley TL. Asparagine 81, an invariant glycosylation site near apyrase conserved region 1, is essential for full enzymatic activity of ecto nucleoside triphosphate diphosphohydrolase 3. Arch Biochem Biophys 2003; 413: 107–15.
Basu S, Murphy-Piedmonte DM, Kirley TL. Conserved lysine 79 is important for activity of ecto-nucleoside triphosphate diphosphohydrolase 3 (NTPDase3). Purinergic Signalling 2004; 1: 51–58.
Wang T-F, Ou Y, Guidotti G. The transmembrane domains of ectoapyrase (CD39) affect its enzymatic activity and quaternary structure. J Biol Chem 1998; 273: 24814–21.
Basu S, Kirley TL. Identification of a tyrosine residue responsible for N-acetylimidazole-induced increase of activity of ecto-nucleoside triphosphate diphosphohydrolase 3. Purinergic Signalling 2005; 1: 271–80.
Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 1989; 56: 777–83.
Ivanenkov VV, Meller J, Kirley TL. Characterization of disulfide bonds in human nucleoside triphosphate diphosphohydrolase 3 (ntpdase3): Implications for NTPDase structural modeling. Biochemistry 2005; 44: 8998–9012.
Ivanenkov VV, Murphy-Piedmonte DM, Kirley TL. Bacterial expression, characterization, and disulfide bond determination of soluble human NTPDase6 (CD39L2) nucleotidase: Implications for structure and function. Biochemistry 2003; 42: 11726–35.
Grinthal A, Guidotti G. Substitution of His59 converts CD39 apyrase into an ADPase in a quaternary structure dependent manner. Biochemistry 2000; 39: 9–16.
Grinthal A, Guidotti G. Transmembrane domains confer different substrate specificities and adenosine diphosphate hydrolysis mechanisms on CD39, CD39L1, and chimeras. Biochemistry 2002; 41: 1947–56.
Wu JJ, Choi LE, Guidotti G. N-linked oligosaccharides affect the enzymatic activity of CD39: Diverse interactions between seven N-linked glycosylation sites. Mol Biol Cell 2005; 16: 1661–72.
Zhong X, Kriz R, Kumar R et al. Distinctive roles of endoplasmic reticulum and golgi glycosylation in functional surface expression of mammalian E-NTPDase1, CD39. Biochim Biophys Acta 2005; 1723: 143–50.
Drosopoulos JH, Broekman MJ, Islam N et al. Site-directed mutagenesis of human endothelial cell ecto-ADPase/soluble CD39: Requirement of glutamate 174 and serine 218 for enzyme activity and inhibition of platelet recruitment. Biochemistry 2000; 39: 6936–43.
Drosopoulos J. Roles of Asp54 and Asp213 in Ca(2+) utilization by soluble human CD39/ecto-nucleotidase. Arch Biochem Biophys 2002; 406: 85.
Kristensen O, Laurberg M, Liljas A et al. Structural characterization of the stringent response related exopolyphosphatase/guanosine pentaphosphate phosphohydrolase protein family. Biochemistry 2004; 43: 8894–900.
Mateo J, Kreda S, Henry CE et al. Requirement of Cys399 for processing of the human ecto-ATPase (NTPDase2) and its implications for determination of the activities of splice variants of the enzyme. J Biol Chem 2003; 278: 39960–68.
Eisenberg D, Luthy R, Bowie JU. VERIFY3D: Assessment of protein models with three-dimensional profiles. A method to identify protein sequences that fold into a known three-dimensional structure. Methods Enzymol 1997; 277: 396–404.
Shi J, Blundell TL, Mizuguchi K. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 2001; 310: 243–7.
Meller J, Elber R. Linear programming optimization and a double statistical filter for protein threading protocols. Proteins 2001; 45: 241–61.
Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci USA 1992; 89: 7290–4.
Schuler H. ATPase activity and conformational changes in the regulation of actin. Biochim Biophys Acta 2001; 1549: 137–47.
Acknowledgement
This work was supported by NIH grants HL59915 and HL72882 to T.L.K.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kirley, T.L., Crawford, P.A. & Smith, T.M. The structure of the nucleoside triphosphate diphosphohydrolases (NTPDases) as revealed by mutagenic and computational modeling analyses. Purinergic Signalling 2, 379–389 (2006). https://doi.org/10.1007/s11302-005-5301-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-005-5301-6