
Abstract
Although the genetic structures of populations in several model organisms have been studied even at the genomic level, quite a few ecologically important or evolutionary interesting species, such as endemic species on oceanic islands, exist whose genetic variations have not yet been studied. Genetic studies of those species may add new insights to our knowledge of evolution, especially when accompanied with ecological and geological knowledge of the species. In this study, we analyzed the genetic variation of two related species of Hamamelidaceae, Distylium racemosum and Distylium lepidotum, living in different habitats and possessing distinctive morphological characteristics. Distylium racemosum is one of the dominant trees of broad-leaved evergreen forests in Japan, and D. lepidotum is a dominant shrub in dry scrub endemic to the Ogasawara Islands. We analyzed the nucleotide variation at 112 protein-coding loci in 95 samples for the two Distylium species and inferred population structure and demographic history on the basis of these data. Our results showed that the samples from two Distylium species were genetically clustered into the following three groups: D. racemosum, D. lepidotum in the Chichijima Island, and D. lepidotum in the Hahajima Island. Furthermore, D. racemosum appears to have diverged first approximately 10 million years ago (MYA), and, then, the split of two populations of D. lepidotum occurred around 1 MYA. Additionally, we detected a few candidate loci that may contribute to adaptation of the species or local populations by exploring the pattern of the variation within and between species using the FST-outlier approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species endemic to oceanic islands are of evolutionary interest because they are thought to have originated from their continental relatives whose habitats are quite different from those of the oceanic island species. Such drastic changes in their habitats will cause alterations of genetic structure of the populations, some of which contribute to adaptation of the species to the new island environments. As a first step to understand the evolutionary process of the formation of oceanic island species, comparisons of the levels of genetic diversity in the oceanic island species with those of the continental source species have been carried out (Stuessy et al. 2014). Although many studies showed populations of oceanic island species harbor lower levels of genetic diversity within population as a result of small population size expected in the island (Crawford et al. 1992; DeJoode and Wendel 1992; Elisens 1992; Frankham 1997; Ito et al. 1998), some population genetic analyses have shown similar or even higher levels of genetic variation in some island taxa compared to those in related continental taxa (i.e., Francisco-Ortega et al. 2000; Chiang et al. 2006; Su et al. 2010; García-Verdugo et al. 2015). The reason for these still remains obscure, but it might be associated with a demographic history (e.g., multiple colonization from mainland, severe bottleneck in continental taxa) and/or changes in ecological and life characteristics (e.g., breeding systems, dispersal ability) (Chiang et al. 2006; Su, Ting and Deng, 2010; García-Verdugo et al. 2015). Comparative population genetic analyses, which include inference on population history, of organisms living in oceanic islands and their close relatives of continental taxa are necessary to understand the evolutionary pattern and processes of the species on oceanic islands.
In the present study, we investigated the genetic variation of a shade-tolerant tree species, Distylium racemosum Sieb. & Zucc. and its close relative, D. lepidotum Nakai, both belonging to the family Hamamelidaceae. Distylium racemosum grows in evergreen broad-leaved forests and is distributed in western Japan, the Ryukyu Islands, Taiwan, Korea, and southern China (Ohba 2001). It is one of the dominant canopy shade-tolerant tree species in pristine evergreen broad-leaved forests in Japan and currently has the highest density in a few such forests (Tanouchi and Yamamoto 1995; Manabe et al. 2000). Dominant shade-tolerant tree species have larger actual population sizes and are thus expected to have larger effective sizes. Additionally, their populations seem less vulnerable to fluctuations during the course of transitions because they constitute climax forests, as compared with shade-intolerant tree species that appear only in the early phases of transition and in occasionally formed gaps.
Its close relative, D. lepidotum, is endemic to the Ogasawara (Bonin) Islands, located in the northwestern Pacific approximately 1000 km south of Tokyo. This species is a dominant tree in dry scrub there (Shimizu and Tabata 1991). However, because D. lepidotum grows on small islands (the largest of which is Chichijima with an area of 24 km2) separated from each other by sea, it may be divided into smaller isolated populations.
Although the two species are closely related, their habitats and morphologies are quite different. While D. racemosum grows up to 20 m or more in height and is distributed in East Asian broad-leaved evergreen forests where humidity is fairly high in summer, D. lepidotum is adapted to dry environments in oceanic islands. Therefore, these species may show genetic traces of adaptation to their habitats. Surprisingly, no studies have yet been conducted on the genetic variation of these two ecologically important species except for a study using microsatellites by Sugai and Setsuko (2016) who uncovered the genetic structure of D. lepidotum. Therefore, even the genetic variation levels as measured by nucleotide diversity have not been known for these two species.
Here, we investigated the genetic variation of these two ecologically important tree species by sequencing 140 loci in 95 individuals sampled from populations of D. racemosum and D. lepidotum, along with one individual from an outgroup species, Hamamelis japonica (Hamamelidaceae). We used targeted amplicon sequencing with next-generation sequencing (NGS) (Bybee et al. 2011) for determining the sequences of these samples. By analyzing the obtained data with population genetic tools, we inferred (1) how the populations of these two species are structured, (2) how much variation exists within and between populations, (3) how these variations have been shaped through past demographic history, and (4) whether there are any loci that may have contributed to adaptive differentiation between the two species or populations within species.
Materials and methods
Samples used in this study
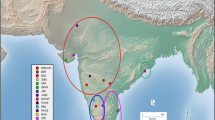
In total, leaf samples from 55 individuals of D. racemosum from 6 sites, Tsushima Islands (TU), Fukuoka (FK), Kagoshima (KG), Miyazaki (MZ) on Kyushu Island, and Ichinomata (IC), Sadayama (SD) on Shikoku Island, and 40 individuals of D. lepidotum from 2 sites, Chichijima Island (CH) and Hahajima Island (HA), were collected. Figure 1 shows locations of the sampling sites and the number of individuals at each location. Additionally, a leaf sample of Hamamelis japonica (HJ) was obtained from Fukuoka in Kyusyu to be used as an outgroup. Genomic DNA was extracted from the leaves using a modified CATB method (Murray and Thompson 1980) and stored at − 30 °C.

Locations of the eight populations surveyed in this study. The number in parentheses indicates the number of samples at each location
Design of primers for amplifying loci and reference sequences
We determined cDNA sequences of D. racemosum by RNA-seq with 454 GS-FLX platform (Roche, Branford, CT, USA). Then, we designed one primer pair for each randomly selected cDNA sequence using the NCBI tool Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). We designed these so that the lengths of the amplified fragments (amplicons) were between 400 and 700 bp. In total, we used 143 primer pairs for PCR amplification in the subsequent NGS analysis. Table S1 shows the primer information.
For obtaining reliable reference sequences to map reads from NGS, we determined the sequence of amplicons (including introns) in one of the samples of D. racemosum using the Sanger method by direct sequencing. When indel polymorphisms hindered the determination of the sequences, we obtained several clones and sequenced each of them.
Amplification of all samples and NGS
We used an Access Array™ System with a 48.48 Access Array IFC (Fluidigm, CA, USA) for performing PCR for the 96 samples. This equipment simultaneously performs multiplexed PCR of 48 different types of amplicons for each of 48 individual DNA samples, saving time and reducing errors that may occur by performing numerous PCRs.
An Agencourt AMPure XP PCR purification kit (Beckman Coulter, Inc., CA, USA) was used to purify the PCR products. The concentration of purified PCR products was measured using a Quantus Fluorometer and QuantiFluor dsDNA system (Promega, WI, USA) and adjusted to 0.2 ng/mol for library construction. The indexed paired-end library for each sample was constructed using a Nextera XT DNA sample prep kit (Illumina, CA, USA). Next, we mixed all the indexed libraries and sequenced them using a MiSeq System (Illumina) with a MiSeq Reagent Kit ver. 3 (600 cycles) with the paired-end (300 + 300) option.
Quality control for raw reads, mapping to reference sequences, SNP calling, and error rate estimation
First, we analyzed quality scores (Q scores) of raw reads from the MiSeq output by applying FastQC v.0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Using fastq_quality_trimmer of FASTX Toolkit 0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit), we trimmed nucleotides of each read with Q score ≤ 25, starting from the end of the read. Next, we removed reads whose lengths were ≤ 50 bp and those < 90% of whose nucleotides had Q score ≤ 25. Next, we used cmpfastq_pe (http://compbio.brc.iop.kcl.ac.uk/software/cmpfastq_pe.php) for classifying paired and unpaired reads. In this procedure, we noticed that the number of forward reads was much higher than that of the reverse reads in almost all samples. Somehow, the qualities of the reverse reads were low. Therefore, we decided to use only the forward reads in the following analyses.
We mapped the reads of each sample to the reference sequence and created SAM files using BWA-MEM (Li and Durbin 2009), which were converted to BAM files using fixmate of SAMtools ver. 1.5 (Li et al. 2009). Thereafter, the BAM files were processed using SAMtools ver. 1.5. First, the BAM files of all the samples were sorted and indexed using sort and index, respectively. Next, using mpileup, the files were converted to the pileup format, which were used for variant call by call in BCFtools with –m option for obtaining VCF files of respective samples.
On the basis of the information obtained from the VCF files, we removed one sample collected from FK in which more than half of the loci had an average read depth of no more than 10. Also, 21 loci with average depth of no more than 10 in 20% or more of the samples were removed. Next, sites with low sums of allelic depths (AD) and quality score of variant call (QUAL) were filtered using bcftools filter. These sites were considered as missing data and replaced with “N” in the FASTA files. Various AD and QUAL threshold values were tried for finding the best filtering in terms of error rates and the number of usable sites.
Lastly, for removing possible duplicated loci, deviations from Hardy–Weinberg equilibrium (HWE) were tested for each of the three groups of populations, identified by a principal component analysis (PCA) described later, using hardy of VCFtools v.0.1.14 (Danecek et al. 2011). A locus was considered duplicated if it contained one or more sites with a significant heterozygote excess in either one of the groups (false discovery rate (FDR) < 0.05). Because we could not clearly determine whether there were two or more duplicated loci and because nucleotide variations at duplicated loci may be affected by gene conversion, we removed all loci suspected to be duplicated from the later analyses. Furthermore, loci were removed when more than two haplotypes were found in the sample whose sequences were determined by the Sanger method because the number of haplotypes must be less than three unless the locus was duplicated.
For estimating the error rate of our NGS sequencing, we compared the sequences obtained by NGS with those obtained by the Sanger method of the sample whose sequences were determined by both methods. The error rates of NGS were described as per site differences between the sequences obtained by the Sanger method and those obtained by NGS. As mentioned above, we changed AD and QUAL values for obtaining optimal filtering with low error rates.
Population genetic analyses
First, for identifying the population structure of all samples, we conducted PCA using TASSEL 5.2.40 (Bradbury et al. 2007) separately for the whole sample, D. racemosum, and D. lepidotum, respectively. In this analysis, sites with missing data were not used. Additionally, we performed cluster analyses using STRUCTURE Version 2.3.2 (Pritchard et al. 2000; Falush et al. 2003) for the data of three sets of the samples using one random site per locus assuming the admixture model. Ten runs for each value of K with 50,000 Markov chain Monte Carlo (MCMC) and 50,000 burn-in iterations were executed. We used CLUMPAK (Kopelman et al. 2015) for summarizing the data for each K and STRUCTURE HARVESTER (Earl and von Holdt 2012) for calculating the mean log-likelihood (Pritchard et al. 2000) and Evanno’s ΔK (Evanno et al. 2005).
Next, we estimated nucleotide diversity between populations (πB), within populations (πW), and FST from the FASTA files using our custom scripts (Supplementary material 1). In this calculation, we computed number of different nucleotides and the average divergence per site for each pair and then computed the average divergence over all pairs. For the groups of populations identified by PCA (CH, HA, and DR groups defined in the “Results” section), we also computed those statistics separately for nonsynonymous and synonymous sites using MEGA7 ver. 7.0 for bigger datasets (Kumar et al. 2016).
For identifying candidate sites under selection, we used BayeScan 2.01 (Foll and Gaggiotti 2008) for all filtered loci including those with some missing data. BayeScan identifies candidate sites under selection by computing the posterior distribution of the site-specific component of FST. Sites under selection were detected with FDR < 0.05. We excluded singleton sites from the data as recommended in the manual. We used the default values for the MCMC algorithm parameters in the BayeScan runs and the prior odds were set to 10. Next, we computed Tajima’s D (Tajima 1989) using the R package PopGenome (Pfeifer et al. 2014). The probability of Tajima’s D value was computed using a custom-made script (Supplementary material 1) that calls a coalescent simulation program ms and its output processing program sample_stats (Hudson 2002). We used an R program, p.adjust, for finding significant loci with FDR < 0.05.
Lastly, to infer the demographic history of the groups of populations (CH, HA, and DR) identified by the PCA analysis, we used fastsimcoal2 2.6 (Excoffier et al. 2013). This program uses two-dimensional joint frequency spectra of populations as input data and searches parameters that maximize the composite likelihood of the data using an efficient algorithm. First, we removed data of the loci identified as under selection by BayeScan or Tajima’s D, because fastsimcoal2 assumes sites evolve neutrally. Furthermore, because this program does not allow missing data at each site, we chose 40 samples from DR (D. racemosum) and 15 samples each from CH and HA with larger numbers of usable sites for this analysis. We used the site frequency spectra of the minor alleles (–m option), since we could not determine the ancestral nucleotide at a significant proportion of loci.
We assumed three demographic models with different migration patterns (No-mig, Is-mig, and Full-mig models; Fig. 2). The No-mig model assumes that there is no migration between the populations. The Is-mig model assumes migration between CH and the HA after the splits of these populations. The Full-mig model assumes migration among all populations after each split. The migration rate was assumed to be symmetric. In all models, population sizes were assumed to change exponentially after the splits. For each demographic model 50 independent runs with broad search ranges for parameters were performed. Each run consisted of 100,000 simulations with 40 ECM cycles. The relative fit of each of the different demographic models to the data was evaluated by means of the Akaike information criterion (AIC) after transforming the log10-likelihood values to ln-likelihoods.

Demographic models used to infer past population structure using fastsimcoal2
For estimating population sizes and the split times of populations in years, we needed the mutation rate per site per generation. We tentatively assumed a generation time of 50 years, considering the age at maturity of the species. The mutation rate, ug, per site per generation was estimated in the following way: Xie et al. (2010) estimated the divergence time between Hamamelis and Distylium to be 51.2 million years ago (MYA) on the basis of six plastid and two nuclear genes and fossil calibration. The estimate of divergence (πB) between Hamamelis and Distylium in the gene set used here was 0.020. Therefore, we estimated ug ≈ 50 × 0.020 / (2 × 5 × 107) = 1.0 × 10−8. Because the mutation rate may have a critical impact on time scaling, we re-estimated the parameters of the best model using a generation time of 100 years (2.0 × 10−8). The recombination rate was assumed to be zero. The confidence interval (CI) of the estimates of the best-fit model was calculated by parametric bootstrapping. Bootstrap replicates (100 replications) were obtained by simulating the site frequency spectra of the minor alleles using the maximum likelihood point estimates (MLE) of the parameters and the same mutation and recombination rates followed by re-estimating the parameters by fastsimcoal2. Finally, to examine the fit of the model to the observed data, we generated simulated sequence data under the model with the MLE for 500 replications using fastsimcoal2, then computed πW, FST, and Tajima’s D and obtained distributions of those statistics using a custom script (Supplementary material 1). These were compared with the observed data.
Results
Quality control of the NGS data and error rate estimation
After initial cleaning of the raw data, the numbers of samples and loci were reduced to 95 and 122, respectively. Significant deviations from HWE or having more than two haplotypes in the sample whose sequences were determined by the Sanger method excluded an additional 10 loci, reducing the total number to 112. Next, we tried various threshold values of AD and QUAL and found that if we set QUAL > 25 and AD > 10, the error rate was 0.025% with 56,573 sites available for the analysis. When we increased the threshold values, the number of usable sites drastically decreased. Thus, we decided to use these threshold values and the sites in some of the samples with lower values were treated as missing data.
Population structure and variation within and between populations
A PCA for 95 samples, including the H. japonica sample, showed that samples of each species (H. japonica, D. racemosum, and D. lepidotum) were clustered together (data not shown). Because H. japonica was distantly separated from the two Distylium species, we conducted a PCA for 94 samples excluding that of H. japonica. Figure 3 shows axes of the first and second principle components. The following three genetic groups were recognized: CH, HA, and D. racemosum, the last of which we hereafter call DR, consisting of TU, FK, IC, SD, MZ, and KG. For examining whether the DR group could be further subdivided or not, we conducted a PCA for the samples from the DR group populations only. However, no clear separation of samples regarding sampling locations was recognized (data not shown). Results from the STRUCTURE analyses were similar (Fig. S1). Therefore, we considered our samples to consist of three groups, CH, HA, and DR. CH and HA were of D. lepidotum and DR was of D. racemosum, and further analyses were mostly conducted on the basis of this classification.

Result of principal component analysis for D. racemosum and D. lepidotum. The first and second principle components are plotted. The populations are indicated by color as explained in the upper inset
Table 1 shows estimates of nucleotide diversity within groups (πW) and between groups (πB) along with FST. The DR group had a significantly higher πW (0.00455) than the CH and HA groups of D. lepidotum (0.00273 and 0.00259, respectively). Although πW in CH was higher than that in HA, the difference was not significant. While only a single individual was sampled from H. japonica, we could still estimate πW assuming random mating. The estimate of πW for H. japonica (0.00288) was smaller than that of D. racemosum and similar to that of D. lepidotum. The divergence between Distylium and Hamamelis measured in πB was estimated to be 0.02. Estimates of FST were around 0.3 between D. racemosum and D. lepidotum and twice that between the two populations of D. lepidotum. Although the populations of D. racemosum were not separately clustered in either the PCA or STRUCTURE analysis, we computed πW, πB, and FST for these (Table S2). Estimates of πW were similar and not significantly different from each other. Estimates of FST were 0.0074–0.0461 and generally low though significantly larger than zero. Therefore, the estimates of πW in respective populations were close to that of the DR group.
We also estimated πW and πB separately for synonymous and nonsynonymous sites using MEGA7 (Table 2). Generally, πW and divergence at synonymous sites were 5–6 times and 4 times larger, respectively, than those at nonsynonymous sites. The estimate of πW in DR was 0.01185 and much larger than those in CH and HA. The synonymous divergence between Distylium and Hamamelis was 0.047–0.048.
Identification of candidate loci under natural selection
We applied BayeScan for the following three datasets of groups: (A) CH, HA, and DR; (B) CH + HA and DR; (C) CH and HA. We found 7, 0, and 1 candidate sites from 4 loci under natural selection, respectively, from datasets A, B, and C with FDR < 0.05 (Fig. S2). Two sites (both nonsynonymous) in the Dr0068 locus, four sites (two nonsynonymous and two synonymous) in the Dr0086 locus, and one site (synonymous) in the Kame143 locus in the dataset A and one site (synonymous) in the Dr0080 locus in the dataset C were identified as candidate sites under natural selection. An examination of the allele frequencies at the sites identified in the dataset A consisting of the data of the three groups showed that the differentiations were between DR and CH + HA (data not shown). Therefore, these sites were differentiated between D. racemosum and D. lepidotum. The site in the Dr0080 locus was differentiated between CH and HA within D. lepidotum. Tajima’s D was not significant at any loci.
Inferring demographic history of populations using fastsimcoal2
For the inference, 4 loci were removed because of suspected action of selection. Furthermore, 7 loci had no site without missing data even though the number of samples was reduced. Thus, 42,437 sites in 101 loci in 40 (DR), 15 (CH), and 15 (HA) samples were used to obtain two-dimensional joint frequency spectra of populations as input data for fastsimcoal2. We tested three different models to evaluate intra- and inter-species gene flow. One model without migration (No-mig model) and two models with migration (Is-mig and Full-mig models) were evaluated. Table 3 showed ln-likelihood and the delta AIC values for each model. Among the three models, Full-mig model had the highest AIC value. This result indicated that there was some intra- and inter-species gene flow after each split. Table 4 shows MLE of the parameters of the best-fit model along with their 95% CI. The split time (t2) between the group DR (D. racemosum) and the two groups CH and HA (D. lepidotum) was estimated to be 11.2 MYA, with a 95% CI of 5.8–17.7 MYA. We estimated that the CH and HA groups of D. lepidotum split from each other 0.9 MYA (t1) with a 95% CI of 0.6–2.1 MYA. When we assumed generation time of 100 years (ug ≈ 2.0 × 10−8), t2 and t1 were estimated to be 6.7 MYA and 0.55 MYA, respectively. Although these estimates were ca. 60% of those estimated by assuming generation time of 50 years, they still indicated that several million years have passed after the split of the two species. After the splits, there was little gene flow between the groups except between CH and HA. The sizes of the ancestral populations (NA1, NA2) were estimated to be 80,000–90,000 when ug ≈ 1.0 × 10−8 and 50,000–60,000 when ug ≈ 2.0 × 10−8. Estimates of population sizes assuming generation time of 100 years were 40–60% of those assuming generation time of 50 years as expected. Both DR and CH expanded, and their current sizes were more than five times larger than the ancestral populations. On the other hand, HA showed population size shrinkage, its current size was more than two times smaller than that of the ancestral population. These results were compatible with the observed values of Tajima’s D, which showed negative values in DR and CH (− 0.788 and − 0.456, respectively; see Fig. S3) while HA showed a positive value (0.233).
Figure S3 shows results examining the fit of the model to the observed values of πW, FST, and Tajima’s D. All observed values were within the 95% interval except for FST between CH and HA (P = 0.040). However, if we assume independence between the statistics, the significance disappears after Bonferroni correction because we had nine observations.
Discussion
In the present study, we found low levels of differentiation between populations of D. racemosum but high differentiation between the two populations of D. lepidotum. Silent nucleotide diversity in D. racemosum as a measure of genetic diversity was fairly high compared with other tree species. We found four candidate genes suspected to have undergone local adaptation between the two species or within D. lepidotum. Lastly, we estimated demographic history by assuming a generation time of 50 years, finding that the two species split 11 MYA and that two island populations of D. lepidotum split 0.9 MYA without much gene flow afterward. Additionally, population expansion seemed to have occurred in populations of D. racemosum and one of the D. lepidotum populations.
Population structure and nucleotide diversity of the two species
The PCA showed that D. racemosum populations in Japan were genetically clustered into one group. This was confirmed by the low estimates (0.0074–0.0472) of FST between these populations. A closer examination of Table S2 reveals that estimates of FST were higher if the SD population was involved in the comparison and that those between the remaining populations were less than 0.02. The reason for higher estimates of FST involving SD is likely the low value of πW in SD, probably caused by a recent bottleneck, not high values of πB to the other populations. Therefore, FST between populations may be mostly lower than 0.02 in this species in Japan. In general, plant species that are long-lived perennial, are outcrossing, or have pollen or seeds dispersed by wind tend to have low levels of genetic differentiation between populations (Hamrick and Godt 1996; Duminil et al. 2007). Although there is little information about the pollination system of D. racemosum, it is thought to be wind pollinated because several genera of Hamamelidaceae with apetalous flowers, large anthers, and stigmas (including Distylium) are probably mainly wind pollinated (Bogle and Philbrich 1980). But exceptions have also been reported in D. lepidotum and two closely related genera, Parrotiopsis and Fothergilla, known to be insect pollinated (Bogle and Philbrich 1980; Abe 2006). Two other dominant canopy tree species in Japanese evergreen broad-leaved forests, Castanopsis sieboldii and C. cuspidata, belonging to Fagaceae but pollinated by insects, show higher levels of differentiation between populations (GST = 0.069 in C. sieboldii and GST = 0.057 in C. cuspidata; Aoki et al. 2014), and Zanthoxylum ailanthoides, a shade-intolerant tree species growing in evergreen broad-leaved forests, showed even high levels of differentiation (FST = 0.13–0.49; Yoshida et al. 2014).
Although it remains possible that D. racemosum is not primarily wind pollinated (and dispersal is not the only factor), wind pollination could be responsible for the low levels of genetic differentiation. Another reason contributing to low differentiation may be that D. racemosum grows in evergreen broad-leaved forests and our samples came from those forests in western Japan. A refugium of evergreen broad-leaved forests in Japan in the last glacial maximum (LGM) is believed to have been in the southern part of Kyushu (Tsukada 1985) and the current populations of D. racemosum in Japan may have expanded from this refugium after the LGM. Therefore, high levels of gene flow between populations due to wind pollination, along with fairly recent splits of the populations and large population size (discussed later), could result in weak differentiation among populations in this species.
Conversely, the samples of the two island populations of D. lepidotum were clustered into two groups each corresponding to one population and the two populations were moderately differentiated (FST = 0.137), which agreed with the results of Sugai and Setsuko (2016), though the absolute values of the estimates differed from each other (FST = 0.0971 in the latter study). Because Hahajima Island is separated from Chichijima Island by about 50 km of seawater and D. lepidotum is insect pollinated, the gene flow rate between the two populations, even by pollen, might be low. This may explain the genetic clustering of the samples and the moderate level of differentiation between the two populations.
Levels of diversity at synonymous sites within populations
Because nucleotide diversity at synonymous sites is generally under much weaker negative selection pressure than at nonsynonymous sites, its level is considered a measure of neutral genetic diversity of the species, with its expected value being 4Neu (Kimura 1969), where Ne and u are effective size and mutation rate, respectively, if we ignore migration. We presumed that Ne would be large in both species because both species are dominant shade-tolerant species in their respective forests, though island-endemic species would have smaller sizes than their mainland counterparts (Frankham 1997). Nucleotide diversity is also proportional to the neutral mutation rate u. Since the divergence rate at synonymous sites between Distylium and Hamamelis was approximately 0.05 (see Table 2) and the split time between these two genera are estimated as ca. 50 MYA (Xie et al. 2010), the mutation rate per year was estimated to be 5 × 10−10 (assuming neutrality at synonymous sites). This rate was much smaller than estimated for angiosperms (the mean being 5.35 × 10−9) and within the range of estimates in gymnosperms (the mean being 7.71 × 10−10; see De La Torre et al. 2017).
As presumed from its dominant status in the forests, even with a lower mutation rate (as indicated by the lower divergence rate per year), the average nucleotide diversity at synonymous sites in D. racemosum was 0.0118, which is in the higher range for tree species, including poplar and pine species (Savolainen and Pyhäjärvi 2007). On the contrary, the average nucleotide diversity at synonymous sites in D. lepidotum was around 0.0065 (55% that of D. racemosum) in each population, in the midrange of other tree species (Savolainen and Pyhäjärvi 2007). Thus, endemicity to small islands might have reduced genetic diversity in D. lepidotum, as shown in other endemic plant species on the Ogasawara Islands (Ito et al. 1998), but it still maintains a fair amount of diversity. Additionally, as shown in our analysis using fastsimcoal2, the estimated population size of CH was rather large (and similar to that of DR), though that of HA was small. This might reflect the size of habitat of D. lepidotum in each of the islands. Distylium lepidotum is dominant in Distylium-Schima dry forests and Distylium-Pouteria dry scrubs in both islands. Such areas occupy ~ 8.3 % of the total area of the Chichijima Island (Shimizu and Tabata 1991); however, it is restricted to small areas in the Hahajima Islands (Shimizu 1994). Thus, lower genetic diversity of the HA population could be due to smaller areas of the island habitats.
Candidate genes under natural selection
The analysis using BayeScan identified four candidate genes under divergent selection, among which three (Dr0068, Dr0086, and Kame143) were differentiated between D. racemosum and D. lepidotum and one (Dr0080) was differentiated between the two island populations of D. lepidotum.
A blast search found that Dr0068 showed similarity to a predicted IQ-DOMAIN (IQD) 31 protein of Vitis vinifera. The IQD protein family in Arabidopsis is known to be involved in regulation of cell function, shape, and growth during plant development thorough interaction with calmodulin and microtubules (Bürstenbinder et al. 2017). The two species in our study have different heights and leaf morphologies along with very different habitats. Two candidate sites possibly under selection underwent a nonsynonymous change, suggesting that differentiation at those sites may cause changes in interaction with either calmodulin or microtubules, leading to morphological differences between species.
Dr0086 showed similarity to At1g51570 in Arabidopsis thaliana, which encodes MCTP4, one of whose functions is in the development of shoot apical stem (Liu et al. 2018a, 2018b). Thus, this gene might be involved in dwarfing of D. lepidotum. Because two of the four candidate sites underwent nonsynonymous substitutions, these might be causative changes for morphological differentiation.
Kame143 showed similarity to At3g52370, encoding fasciclin-like arabinogalactan protein 15 (FLA15). Members of the FLA gene family in Arabidopsis have a conserved domain important for cell adhesion (Johnson et al. 2003). Although specific functions of FLA15 are to be elucidated yet, the gene might be involved in morphogenesis because some members of the FLA family are known to affect cell wall architecture in Arabidopsis and Eucalyptus (MacMillan et al. 2010).
Dr0080, which was differentiated between the CH and HA populations of D. lepidotum, showed similarity to a predicted protein, PLASTID MOVEMENT IMPAIRED 1 (PMI1), of Quercus suber. PMI1 has been shown to be involved in chloroplast and nuclear photorelocation movements in mesophyll cells (Suetsugu et al. 2015). These movements are necessary for absorbing more blue light when it is weak and for avoiding it when too strong. Therefore, differences in the photo-environments where respective samples (from the Chichijima and Hahajima islands) reside might have caused the differentiation.
Demographic history of the populations of D. racemosum and D. lepidotum
The Ogasawara Islands are oceanic islands 1000 km south of Japan. Four putative sources of the organisms on the islands are (1) southeastern tropical and subtropical Asia, including the Philippines, Taiwan, and the Ryukyu Islands; (2) Japan, especially its southern part; (3) the South Pacific or Polynesian Islands; and (4) the Mariana Islands (Ono 1991). There are 12 Distylium species in eastern Asia (Zhang and Endress 2003), which are candidates of the origin of D. lepidotum. The distributions of most of these species are limited to Southeastern China excepting D. racemosum and D. gracile. Although D. gracile grows both in Southeastern China and Taiwan, it also has a limited distribution. Considering the wide distribution of D. racemosum from western Japan through Ryukyu and Taiwan to South China, the origin of D. lepidotum seems to be D. racemosum or a related species once distributed in one of those places. Our older estimate of the split between the two species was 11.2 MYA (CI, 5.8–17.7 MYA), though this may or may not correspond to the split between the two species, because D. lepidotum may have originated from other populations than the populations of D. racemosum we investigated here (e.g., Taiwan, Ryukyu Islands, and Izu Islands). Chichijima and Hahajima islands were first formed as underwater volcanic islands 48–44 MYA and appeared above water a few million years later (Umino and Nakano 2007; Umino et al. 2016). However, up until 30 MYA, the Shikoku Basin that now separates the Izu–Bonin–Mariana Arc (IBMA), on which the Ogasawara Islands reside, and the Kyushu-Palau Ridge (KPR), closer to southern Kyushu, was not formed and the IBMA and KPR were very close (Okino et al. 1994). The expansion of the Basin took about 15 MYA to complete. It is not clear whether the split between D. racemosum and D. lepidotum occurred during or after the expansion of the Shikoku Basin because the CI of the split overlaps the expansion period of the Basin. However, the chance of immigration to the islands would be higher if they were close to the source populations. Therefore, immigration to the Ogasawara Islands might have occurred during the expansion. After the founding of the population that evolved to D. lepidotum in the Ogasawara Islands, gene flow between the two species has been almost nonexistent. Our result showed that inter-species migration that occurred between the ancestral populations (mDA) was much smaller than those between the present populations of D. lepidotum (mDC and mDH), supporting this hypothesis (Table 4).
The split between the Chichijima and Hahajima populations was estimated to be 1 MYA (CI, 0.64–2.0 MYA) when we assumed generation time of 50 years, and it was 0.55MYA when the generation time was 100 years. The split time corresponds to the Pleistocene in both cases. The level of gene flow between the two populations was estimated to be higher (1.3 × 10−5) than those between D. lepidotum and D. racemosum, which could be explained by the short distance (ca. 50 km) and rather shallow seawater separating them (200 m or less). Ito (1998) proposed that, in the evolution of endemic flora on oceanic islands, there should be four stages in succession: migration, establishment, enlargement, and diversification. Because one candidate locus under divergent selection was identified by our analysis, these two populations may be in the fourth stage (diversification) in the evolution of endemic species of oceanic islands (Ito 1998).
The DR group (D. racemosum) and the Chichijima population (D. lepidotum) have both been expanding after the splits. Their current sizes were estimated to exceed a few hundred thousand, while the Hahajima population (D. lepidotum) was inferred to be shrinking. These findings are compatible with the estimates of the average Tajima’s D across loci (see Fig. S3). Although the time scale of expansion in D. racemosum seems unlikely to be 10 million years (we think this estimation was caused by the small number of parameters in the model), the time scale of these expansions and shrinkage do not correspond to one or a few glacier cycles because such rapid changes would not cause non-zero values of Tajima’s D. Two points may be noted concerning these size changes. First, the large estimates of the current sizes of the DR and Chichijima populations make sense, considering the dominant status of the two species. Second, even though the mean nucleotide diversity was not significantly different between the Chichijima and Hahajima populations of D. lepidotum, their current sizes seem quite different. This may indicate the importance of inferring past population history for understanding the current status of populations.
Conclusion
We investigated genetic structure and evolutionary history of two Distylium species, D. racemosum and D. lepidotum, both dominant tree species in their respective habitat forests in Japan. The two species split about 10 MYA and have exchanged very few migrants afterwards, probably because they were separated by more than 1000 km of seawater. Because of the separation and differences in habitats, morphological and possibly physiological differences evolved between the two species. We found three loci under divergent selection, which might contribute to morphological differences, out of 116 loci investigated. As expected from its dominant status in the forests, D. racemosum maintained a large amount of genetic variation as measured by nucleotide diversity at synonymous sites, even though the per year mutation rate in the species is rather low. One surprising result from our inference of demographic history in the two species was that the size of the Chichijima population was currently large and increasing, while the Hahajima population was relatively small and shrinking. This finding, along with the possible presence of candidate genes under local selection, may have important implications for the conservation of D. lepidotum.
The present analyses of sequence polymorphisms, even though lacking comprehensive sampling of the species range, provides important insights into those ecologically significant, but thus far largely neglected species by evolutionists. Expanded sampling over their entire distribution and sequencing of more loci should uncover further interesting details about the evolutionary history and features of these two species.
References
Abe T (2006) Threatened pollination systems in native flora of the Ogasawara (Bonin) islands. Ann Bot 98:317–334. https://doi.org/10.1093/aob/mcl117
Aoki K, Ueno S, Kamijo T, Setoguchi H, Murakami N, Kato M, Yd T (2014) Genetic differentiation and genetic diversity of Castanopsis (Fagaceae), the dominant tree species in Japanese broadleaved evergreen forests, revealed by analysis of EST-associated microsatellites. PLoS One 9:e87429. https://doi.org/10.1371/journal.pone.0087429
Bogle AL, Philbrich CT (1980) A generic atlas of Hamamelidaceous pollens. Contrib Gray Herb Harvard Univ 210:29–103
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Bybee SM, Bracken-Grissom H, Haynes BD et al (2011) Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biol Evol 3:1312–1323. https://doi.org/10.1093/gbe/evr106
Bürstenbinder K, Möller B, Plötner R, Stamm G, Hause G, Mitra D, Abel S (2017) The IQD family of calmodulin-binding proteins links calcium signaling to microtubules, membrane subdomains, and the nucleus. Plant Physiol 173:1692–1708. https://doi.org/10.1104/pp.16.01743
Chiang Y-C, Hung K-H, Schaal BA et al (2006) Contrasting phylogeographical patterns between mainland and island taxa of the Pinus luchuensis complex. Mol Ecol 15:765–779. https://doi.org/10.1111/j.1365-294X.2005.02833.x
Crawford DJ, Stuessy TF, Haines DW et al (1992) Allozyme diversity within and divergence among four species of Robinsonia (asteraceae: Senecioneae), a genus endemic to the Juan Fernandez Islands, Chile. Am J Bot 79:962–966. https://doi.org/10.1002/j.1537-2197.1992.tb13680.x
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
DeJoode DR, Wendel JF (1992) Genetic diversity and origin of the Hawaiian Islands cotton, Gossypium tomentosum. Am J Bot 79:1311–1319. https://doi.org/10.2307/2445059
De La Torre AR, Li Z, Van de Peer Y, Ingvarsson PK (2017) Contrasting rates of molecular evolution and patterns of selection among gymnosperms and flowering plants. Mol Biol Evol 34:1363–1377. https://doi.org/10.1093/molbev/msx069
Duminil J, Fineschi S, Hampe A et al (2007) Can population genetic structure be predicted from life-history traits? Am Nat 169:662–672. https://doi.org/10.1086/513490
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Elisens WJ (1992) Genetic divergence in Galvezia (Scrophulariaceae): evolutionary and biogeographic relationships among South American and Galápagos species. Am J Bot 79:198–206. https://doi.org/10.1002/j.1537-2197.1992.tb13638.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M (2013) Robust demographic inference from genomic and SNP data. PLoS Genet 9:e1003905. https://doi.org/10.1371/journal.pgen.1003905
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant Mmarkers: a Bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Francisco-Ortega J, Santos-Guerra A, Kim S-C, Crawford DJ (2000) Plant genetic diversity in the Canary Islands: a conservation perspective. Am J Bot 87:909–919. https://doi.org/10.2307/2656988
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327. https://doi.org/10.1038/hdy.1997.46
García-Verdugo C, Sajeva M, Mantia TL et al (2015) Do island plant populations really have lower genetic variation than mainland populations? Effects of selection and distribution range on genetic diversity estimates. Mol Ecol 24:726–741. https://doi.org/10.1111/mec.13060
Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond Ser B Biol Sci 351:1291–1298. https://doi.org/10.1098/rstb.1996.0112
Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18:337–338. https://doi.org/10.1093/bioinformatics/18.2.337
Ito M (1998) Origin and evolution of endemic plants of the Bonin (Ogasawara) Islands. Res Popul Ecol 40:205–212. https://doi.org/10.1007/BF02763405
Ito M, Soejima A, Ono M (1998) Genetic diversity of the endemic plants of the Bonin (Ogasawara) Islands. In: Stuessy TF, Ono M (eds) Evolution and speciation of island plants. Cambridge University Press, Cambridge, pp 141–154
Johnson KL, Jones BJ, Bacic A, Schultz CJ (2003) The fasciclin-like arabinogalactan proteins of Arabidopsis. A multigene family of putative cell adhesion molecules. Plant Physiol 133:1911–1925. https://doi.org/10.1104/pp.103.031237
Kimura M (1969) The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61:893–903
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Liu L, Li C, Liang Z, Yu H (2018a) Characterization of multiple C2 domain and transmembrane region proteins in Arabidopsis. Plant Physiol 176:2119–2132. https://doi.org/10.1104/pp.17.01144
Liu L, Li C, Song S et al (2018b) FTIP-dependent STM trafficking regulates shoot meristem development in Arabidopsis. Cell Rep 23:1879–1890. https://doi.org/10.1016/j.celrep.2018.04.033
MacMillan CP, Mansfield SD, Stachurski ZH, Evans R, Southerton SG (2010) Fasciclin-like arabinogalactan proteins: specialization for stem biomechanics and cell wall architecture in Arabidopsis and Eucalyptus. Plant J 62:689–703. https://doi.org/10.1111/j.1365-313X.2010.04181.x
Manabe T, Nishimura N, Miura M, Yamamoto S (2000) Population structure and spatial patterns for trees in a temperate old-growth evergreen broad-leaved forest in Japan. Plant Ecol 151:181–197. https://doi.org/10.1023/A:1026512404110
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Ohba H (2001) Hamamelidaceae. In: Iwatsuki K, Boufford DE, Ohba H (eds) Flora of Japan, Vol 2b: Archichlamydeae (b). Kodansya, Tokyo, pp 5–9
Okino K, Shimakawa Y, Nagaoka S (1994) Evolution of the Shikoku Basin. J Geomagn Geoelectr 46:463–479. https://doi.org/10.5636/jgg.46.463
Ono M (1991) The flora of the Bonin (Ogasawara) Islands. Aliso 13:95–105. https://doi.org/10.5642/aliso.19911301.04
Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ (2014) PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol 31:1929–1936. https://doi.org/10.1093/molbev/msu136
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Savolainen O, Pyhäjärvi T (2007) Genomic diversity in forest trees. Curr Opin Plant Biol 10:162–167. https://doi.org/10.1016/j.pbi.2007.01.011
Shimizu Y (1994) Ogasawara syotou hahajima rettou no syokusei – kansei teibokurin no bunpu sosei kouzou wo tyuushinnni. Komazawa Geogr 30:17–68 (in Japanese)
Shimizu Y, Tabata H (1991) Forest structures, composition, and distribution on a Pacific Island, with reference to ecological release and speciation. Pac Sci 45:28–49
Stuessy TF, Takayama K, López-Sepúlveda P, Crawford DJ (2014) Interpretation of patterns of genetic variation in endemic plant species of oceanic islands. Bot J Linn Soc 174:276–288. https://doi.org/10.1111/boj.12088
Su Y, Wang T, Deng F (2010) Contrasting genetic variation and differentiation on Hainan Island and the Chinese mainland populations of Dacrycarpus imbricatus (Podocarpaceae). Biochem Syst Ecol 38:576–584. https://doi.org/10.1016/j.bse.2010.07.003
Suetsugu N, Higa T, Kong S-G, Wada M (2015) PLASTID MOVEMENT IMPAIRED1 and PLASTID MOVEMENT IMPAIRED1-RELATED1 mediate photorelocation movements of both chloroplasts and nuclei1. Plant Physiol 169:1155–1167. https://doi.org/10.1104/pp.15.00214
Sugai K, Setsuko S (2016) Novel microsatellite markers for Distylium lepidotum (Hamamelidaceae) endemic to the Ogasawara Islands. BMC Res Notes:9. https://doi.org/10.1186/s13104-016-2137-9
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tanouchi H, Yamamoto S (1995) Structure and regeneration of canopy species in an old-growth evergreen broad-leaved forest in Aya District, southwestern Japan. Vegetatio 117:51–60. https://doi.org/10.1007/BF00033258
Tsukada M (1985) Map of vegetation during the last glacial maximum in Japan. Quat Res 23:369–381. https://doi.org/10.1016/0033-5894(85)90041-9
Umino S, Nakano S (2007) Geology of the Chichijima Retto District. Qadrangle Series, 1:50,0000, Geological Survey of Japan. AIST (in Japanese with English abstract)
Umino S, Ishizuka O, Kanayama K (2016) Geology of the Hahajima Retto District. Quadrangle Series, 1:50,000, Geological Survey of Japan, AIST, 46 p(in Japanese with English abstract 2 p.)
Xie L, Yi T-S, Li R, Li D-Z, Wen J (2010) Evolution and biogeographic diversification of the witch-hazel genus (Hamamelis L., Hamamelidaceae) in the Northern Hemisphere. Mol Phylogenet Evol 56:675–689. https://doi.org/10.1016/j.ympev.2010.02.018
Yoshida T, Tamekuni M, Yahara T, Inomata N, Tachida H (2014) Demographic history of a common pioneer tree, Zanthoxylum ailanthoides, reconstructed using isolation-with-migration model. Tree Genet Genomes 10:1213–1222. https://doi.org/10.1007/s11295-014-0755-8
Zhang H and Endress PK (2003) Hamamelidaceae. In: Wu ZY, Raven PH and Hong DY (eds) Flora of China, Vol 9 Pittosporaceae through Connaraceae, Science Press, Beijing, and Missouri Botanical Garden, St. Louis), pp. 28–30.
Acknowledgments
We thank two anonymous reviewers and the Associate Editor for their thoughtful comments on an earlier version of this manuscript. We thank Atsuya Kamei, Makiko Mimura, Atsushi Sakai, and Naoyuki Nishimura for helping us to collect samples used in this study.
Data archiving statement
The sequences data from this study have been submitted to the DDBJ Sequence Read Archive (SRA) (https://www.ddbj.nig.ac.jp/dra/index.html) under Accession No. DRA008576.
Funding
This research was partially supported by the JSPS, grant numbers JP26291082 (HT), JP16H02553 (HT), and 16K07466 (JK) and 2331067 (SS) and the Environment Research and Technology Development Fund of the Ministry of the Environment, Japan (4-1402, SS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Y. Tsumura
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hitomi Yagi and Jie Xu contributed to this work equally.
Electronic supplementary material
ESM 1
(DOCX 2972 kb)
Rights and permissions
About this article

Cite this article
Yagi, H., Xu, J., Moriguchi, N. et al. Population genetic analysis of two species of Distylium: D. racemosum growing in East Asian evergreen broad-leaved forests and D. lepidotum endemic to the Ogasawara (Bonin) Islands. Tree Genetics & Genomes 15, 77 (2019). https://doi.org/10.1007/s11295-019-1386-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-019-1386-x