
Abstract
During Quaternary glaciations, the ranges of Northern Eurasia forest species periodically experienced contraction followed by subsequent re-colonizations in the interglacial intervals. However, unlike the broadleaf trees of temperate forests, taiga species seem not to have retreated fully to southern regions in unfavorable periods and possibly survived at mid-latitudes in multiple refugia. Here, we report a study of genetic variation of three mitochondrial DNA markers in 90 populations of Scots pine (Pinus sylvestris) located from Eastern Europe to Eastern Siberia. The geographic distribution of seven mitotypes demonstrated the split between western and eastern populations approximately along the 38th meridian. Genetic diversity in the western part was significantly higher than in the eastern one. Five mitotypes were western- and one eastern-specific. One mitotype was common in both regions, but in the eastern part it occurred only in the South Urals and adjacent areas. The geographic structure in the mitotype distribution supports a hypothesis of post-glacial re-colonization of the studied territory from the European and Ural refugia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pleistocene glaciations caused fundamental rearrangements of communities of living organisms. Species of forest biota reduced their ranges in the glacial phases, surviving in isolated refugia and expanding under climate improvement. Demographic processes associated with these events have left traces in the level and structure of genetic variation, and the analysis of which, combined with the paleontological data, makes it possible to explore the history of the species.
Scots pine is one of the keystone species of the forest landscape in Europe and North Asia. According to paleodata during the last ice age, unlike many thermophilic species, Scots pine survived not only in the Mediterranean refugia but also in Central Europe, Hungary, Slovakia (Willis and van Andel 2004), and the Carpathian region (Haesaerts et al. 2010). In Siberia, Scots pine macrofossils corresponding to the last glaciation were found in the Low Irtysh area (Binney et al. 2009). Range-wide population studies showed a trend of reduction of genetic diversity in Scots pine populations toward the east that favors the assumption of European origin of modern pine populations in the taiga zone of Eurasia. For example, the mean number of allozyme alleles per locus in the Amur basin (East Asia) was less (2.15) than in Central Europe (2.58), although not significantly (Sannikov and Petrova 2012), and the standardized number of haplotypes per population at the chloroplast microsatellite locus r 14-1 was 9.2 in the Asian part of the range vs. 9.7 (P = 0.03) in the European Russia (Semerikov et al. 2014).
Expanding east after one of the major glaciations, Scots pine populations likely experienced genetic drift, accompanied by a gradual decrease in diversity due to multiple foundation events. However, the time and the sequence of colonization and extinction events are still unclear, as well as whether the refugia of Scots pine existed in the eastern part of the species area, and what role they played in the post-glacial re-colonization.
Mitochondrial genetic markers having maternal inheritance in conifers (family Pinaceae) are transmitted by seeds and thus have a reduced mobility, unlike the paternally inherited chloroplast markers transmitted by pollen and bi-parentally inherited nuclear DNA markers transmitted by both seeds and pollen. As a result, traces of historical demographic events, such as decline, expansion, isolation, and migration of populations are better preserved in the genetic structure of populations identified by such markers. The mitochondrial DNA was widely used in studies of genetic structure and phylogeography of Scots pine (Sinclair et al. 1999; Soranzo et al. 2000; Cheddadi et al. 2006; Naydenov et al. 2007; Pyhäjärvi et al. 2008; Vidyakin et al. 2012; Buchovska et al. 2013; Bernhardsson et al. 2016; Dering et al. 2017). In addition to Mediterranean refugia, which were common for many forest species, the geographical distribution of mitochondrial DNA haplotypes (mitotypes) showed additional potential northern sources of post-glacial re-colonization of Scots pine in northern Europe and Asia (Naydenov et al. 2007; Pyhäjärvi et al. 2008). Data on the geographic distribution of 5 bp deletion in the first intron of the mitochondrial gene nad7 that marks the migration route out of the “northern” refugium demonstrated the lack of this marker to the east from the line, roughly corresponding to North Dvina–Volga Rivers, suggesting the existence of additional glacial refugium to the east of this line (Vidyakin et al. 2012). The likely location of such refugium could be the Ural Mountains, considered as a glacial shelter for forest species (Sannikov and Petrova 2012; Vidyakin et al. 2012). At the same time, the lack of variability of mitochondrial DNA in the eastern part of the P. sylvestris range did not allow to locate more specifically this hypothetical refugium in the previous studies. To identify these possible Pleistocene refugia, we studied the Scots pine variation in 90 population samples collected from Eastern Europe to Transbaikalia using the newly developed mitochondrial DNA markers.
Materials and methods
DNA samples isolated from two to 24 individual trees per population were genotyped in 90 populations (Supplementary material Table S1; Fig. 1). Among them, 36 populations were used previously (Vidyakin et al. 2012; Semerikov et al. 2014), and 52 were newly collected. DNA was isolated using the CTAB method (Devey et al. 1996) from fresh or silica gel dried needles. Three mitochondrial DNA markers polymorphic in the eastern part of the P. sylvestris range were used: nad7 (Naydenov et al. 2007; Pyhäjärvi et al. 2008; Vidyakin et al. 2012; Buchovska et al. 2013), coxI (Semerikov et al. 2015) and a new syn31 marker recently designed based on the whole genome sequencing approach described in Semerikov et al. (2015). In brief, the development of the syn31 marker included the following steps: low coverage paired-end sequencing of the genome of a single Scots pine tree taken from a natural population near Ekaterinburg, Russia, using Illumina HiSeq 2000, assembly of contigs and selection of the contigs homologous to the mitochondrial genome sequences of conifer species. Then, the specific PCR primers were designed for some regions represented by the identified mitochondrial genome sequences and were used to partly re-sequence them and to search for polymorphism in a sample of eight individuals representing different parts of the Scots pine range—Mongolia, northwest Russia, the Carpathians, and the Caucasus.

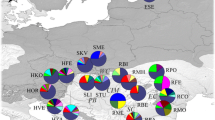
a Network of seven mitotypes found in Pinus sylvestris in Eastern Europe and Northern Asia. Size of the circles is roughly proportional to the frequency of the each mitotype. b Geographic distribution of sampled populations. Approximate locations of the P. sylvestris macrofossils corresponding to the last glaciation (Willis and van Andel 2004; Binney et al. 2009; Haesaerts et al. 2010) are indicated by the star symbol. The mitotype frequencies in each population are represented with pie diagram. Five SAMOVA groups are outlined by tick rose line. See population names in Supplementary material Table S1
The PCR primers GTTGGGTGCAGTAAGGGAAA and TATACCCACCCCAACCAAGA were designed for the syn31 marker using the 12,069-bp-long mitochondrial contig NODE_new_31_length_12111 (GeneBank accession number KY302806) to amplify the 1161-bp-long region (positions 4522–5682 in the contig). Two fragments were detected: A (amplification type A) and C (amplification type C) of the same length (1161 bp). The sequence of the C fragment (KY302807) was identical to the respective region in the contig NODE_new_31_length_12111, but the sequence of fragment A (KY302808) differed from C by 18 SNPs. There was also the third amplification type M in some trees that consisted of two fragments similar to A and C. Based on our segregation data obtained for progeny of six trees (data are not presented), the amplification type C very likely represents a mitochondrial fragment, but origin of the A and M types is unclear, due to possible amplification of nuclear copies of this locus. Therefore, to be conservative and to avoid any misgenotyping errors, we regard this marker as having two mitotypes C and non-C (A or M) in this study.
Variation of the nad7 marker was determined as an indel by presence or absence of a 5-bp-long sequence in the first intron of the nad7 gene. It was genotyped via amplification with the PCR primers described in Jaramillo-Correa et al. (2004). The PCR was performed in a volume of 25 μL, containing 25 ng of genomic DNA, 1× PCR buffer (75 mm Tris-HCl, 20 mM (NH4)2SO4, 0.1% Tween-20), 2.5 mM MgCl2, 200 μM of each dNTP, 0.2 μM forward and reverse primer, 0.32 units of Taq polymerase (SibEnzyme Ltd., Novosibirsk, Russia). The PCR program consisted of an initial denaturation at 94 °C for 5 min and 35 cycles of amplification: 94 °C, 30 s; 60 °C, 45 s; 72 °C, 2 min. A final elongation was 7 min at 72 °C. After restriction with HaeIII, the products were separated in a denaturing polyacrylamide gel followed by silver staining.
Variation of the coxI marker was determined by a single nucleotide substitution (SNP) A/C in the 3′-UTR region of the coxI gene. Genotyping of the coxI gene was carried out by sequencing of the respective PCR product amplified by the PCR primers CCTTCTTTCCCATGCATTTC and CGTGGTACCCAGGAGAAGAA. Sequencing was done using the first primer.
For genotyping of the syn31 marker, the respective fragments were amplified first using the same protocol and the above mentioned primers, then the PCR product was digested with the TaqI restriction enzyme, and resulted product was run in a denaturing acrylamide gel followed by staining with silver nitrate, which finally allowed us to discriminate two mitotypes: “C” and “not-C.” There were seven combinations of all alleles for these three markers that were designated as seven mitotypes, respectively (Table 1). The mitotype network was constructed using NETWORK 5.0.0.1 and median-joining method (Bandelt et al. 1999).
Hierarchical analysis of molecular variation (AMOVA) within populations, among populations within groups and between groups was performed using the Arlequin v.3.5 software (Excoffier et al. 2005). Statistical significance of fixation indexes was assessed by 1000 permutations.
Additional fixation indexes were calculated and compared to each other: G ST (Nei 1987) based only on the frequencies of mitotypes and N ST taking into account the genetic distance between mitotypes (Pons and Petit 1996) using the PermutCpSSR v.1.0 software (Burban et al. 1999), in which algorithm is based on the permutations over mitotypes. Rate of the permutations for which N ST was greater than or equal to the N ST of the real data corresponds to the probability that N ST > G ST was random. It does not matter whether genetic differences between mitotypes have arisen phyletically or by recombination. If N ST > G ST , then genetically related mitotypes tend to coexist in the same population. The clustering of populations was done using the SAMOVA program (Dupanloup et al. 2002), which takes into account both the molecular data and the geographic location of populations. The algorithm of the program is aimed at finding a clustering of geographically adjacent populations into the K groups, in which the differentiation of groups (F CT ) would be the greatest, wherein K is given a priori. The analysis was performed at K = 2, 3, 4, 5, and 6. The geographic distribution of the mitochondrial DNA mitotypes identified in our study was manually drawn, and the distribution of mitotypes that is based on the nad7 and nad1 markers from the data of obtained in Naydenov et al. (2007), Pyhäjärvi et al. (2008), Buchovska et al. (2013), Dering et al. (2017) and our study was constructed using the program PhyloGeoViz (Tsai 2011).
Results
Seven mitotypes were identified in 1132 trees in 90 populations using three mitochondrial loci (Table 1). Their geographical distribution was very heterogeneous (Fig. 1).
The SAMOVA analysis of the spatial structure of genetic variation demonstrated that F CT values reached plateau (0.74) when the number of groups of populations was 5. The largest group I was located in the eastern part of the range, approximately to the east of 38° N, within which there was a small group IV in the southern part of the Urals. To the west of the group I there were groups II (Belorussia, western Russia, and north-east Scandinavia, populations 71, 73, 78, 79, 80, 82, 85, and 88), III (Slovakia and Estonia, populations 81 and 87), and V (central Russia, populations 70, 72, 74, 77, 83, 84, and 86) (Fig. 1). Analysis of the structure of genetic variation using AMOVA (Table 2) based on division of the populations into five SAMOVA groups (Fig. 1) confirmed the significant contribution of the differences between populations to the total diversity of mitochondrial DNA (F ST = 0.77), and the major differences were observed between groups (F CT = 0.74).
The mitotypes 2, 3, and 4 dominated in the western populations (SAMOVA groups II, III, and V), while 1 in the eastern populations (groups I and IV). The eastern and western groups differed greatly also by the level of diversity (Supplementary material Table S1). The average number of mitotypes and mitotype diversity in western populations were 3.60 and 0.558, respectively, and significantly higher than in the eastern ones: 1.3 and 0.080, respectively (P < 0.0001, Mann-Whitney U test).
Mitotype 1, typical for the eastern populations, was also present in the west (Fig. 1), but with decreasing frequency in a western direction, likely due to introgression from the east. However, lack of the data from the Central and Western Europe prevents us from making a more definite conclusion. Most of the mitotypes of the western groups were completely absent in the east. The detected significant excess of N ST over G ST (P = 0.05) confirms the presence of the phylogeographic structure in the mitochondrial DNA variation, which means that the related mitotypes tend to co-occur in the same population.
Distribution of mitotypes within western and eastern parts of the studied area was also uneven. In the east, besides mitotype 1 the mitotype 2 was present, which dominates in the Southern Urals, but gradually disappears to the west and east of this region, however, occurred within the group I as far to the west as in the Chaadaevka population (#51) and as far to the east as in the Surgut population (#39) (Fig. 1; Supplementary material Table S1). Noteworthy that a very mosaic structure was revealed in the densely sampled South Urals, where the frequency of mitotype 2 can drop from 100% to zero within a short distance of just several tens of kilometers, which could be due to strong founder effects. Interestingly, mitotype 2 was widespread in the west. In the western part of the Russian Plain, the endemic to this region mitotype 5, which is probably a product of recombination between mitotypes 1 and 3 was found in populations Ryazan (#74), Tver (#77), Usman (#83), Navlinskoye (#84), and Domgeritsa (#79). Moreover, in Belarus mitotypes 6 and 7 were sporadically found, which were probably also the result of recombination between common mitotypes 1–5.
Discussion
The study of the mtDNA diversity of P. sylvestris revealed a complex history of re-colonization in the studied area between Slovakia on the west and Yakutia on the east. Split of the populations into two groups can be explained by two main sources of re-colonization—eastern and western. The similarities of the mitotype frequencies across the territory from Belarus to the Kola Peninsula supports the recent expansion of the western groups from a single source—a refugium probably located in Carpathian Mountains, which is consistent with the presence of pine trees in Central Europe during the LGM (Willis and van Andel 2004; Haesaerts et al. 2010). The assumption of Buchovska et al. (2013) that Scandinavia was re-colonized from a refugium located to the south-east of Moscow seems less likely, because under the dry and cold climate of glacial epoch, the plains were less suitable for survival of trees, compared to mountain regions. Within the range of eastern groups I and IV, mitotype 1 is almost universally distributed, and only mitotype 2 is confined to a compact region around the Southern Urals (group IV), which indicates its spread from this center. We hypothesize that the colonization of the entire eastern part of the studied area occurred from this area. In this case, the loss of the mitotype 2 on the periphery of group IV can be explained by a sequence of founder events during colonization. Alternatively, the rest of the east group I was colonized from other refugia located outside the Urals, which are not detectable due to lack of the variation of mitochondrial DNA. For more definite conclusions, additional genetic and paleobotanical data are needed, in particular, the additional markers of mitochondrial DNA, which would help us identify the migration routes of Scots pine in the east of the Urals.
This study, as well as the research of Vidyakin et al. (2012) did not find a 5-bp-long deletion in the first intron of the mitochondrial gene nad7 of Scots pine in the eastern part of the range, contrary to Naydenov et al. (2007) and Buchovska et al. (2013), who found that this deletion occurred, sometimes with high frequency, to the east, up to the Baikal region. We guess that this discrepancy may be caused by errors in transfer of seed material across regions used for reforestation. The distribution of the mitotypes based on the data for the nad7 and nad1 markers obtained by Naydenov et al. (2007), Pyhäjärvi et al. (2008), Buchovska et al. (2013), Dering et al. (2017) and combined with data of this study (except doubtful populations) is presented in Fig. S1 (Supplemental data).
In conclusion, it should be noted that a much larger diversity of mitotypes in the western group compared to the eastern one and lack of the mitotypes as well as mtDNA mutations specific exclusively to the eastern group favors the western, probably the Central European, origin of the eastern group. However, the events related to this range expansion took place apparently long before the migrations resulted in the modern structure of Scots pine populations. The presence of the secondary center of the re-colonization under the initially European origin is not a feature unique to Scots pine and was noted for some other species of forest biota of Northern Eurasia, for example, Calluna vulgaris (Sannikov et al. 2014) and Juniperus communis (Hantemirova et al. 2017). Clearly, migration of P. sylvestris (as well as C. vulgaris and J. communis) to the Urals from Central Europe could be related to the restoration of forest zone after one of the most severe glacial intervals, when forest vegetation disappeared completely on the plains of Eastern Europe and Western Siberia and in the mountainous regions of the Urals, for example, during MIS8 (270–250 kya) or MIS6 (130–190 kya), when the boundary of glaciation in Western Siberia reached 60°–62° N, respectively (Volkova et al. 2002). To obtain the historic time estimates of re-colonization of the eastern part of the Scots pine range, more research is needed based on multilocus nuclear data obtained using next-generation sequencing methods.
References
Bandelt H-J, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bernhardsson C, Floran V, Ganea SL, Garcia-Gil MR (2016) Present genetic structure is congruent with the common origin of distant Scots pine populations in its Romanian distribution. For Ecol Manag 361:131–143. https://doi.org/10.1016/j.foreco.2015.10.047
Binney HA, Willis KJ, Edwards ME, Bhagwat SA, Anderson PM, Andreev AA, Blaauw M, Damblon F, Haesaerts P, Kienast F, Kremenetski KV, Krivonogov SK, Lozhkin AV, MacDonald GM, Novenko EY, Oksanen P, Sapelko TV, Valiranta M, Vazhenina L (2009) The distribution of late-quaternary woody taxa in northern Eurasia: evidence from a new macrofossil database. Quat Sci Rev 28(23-24):2445–2464. https://doi.org/10.1016/j.quascirev.2009.04.016
Buchovska J, Danusevicius D, Baniulis D, Stanys V, Siksnianiene JB, Kavaliauskas D (2013) The location of the northern glacial refugium of Scots pine based on mitochondrial DNA markers. Balt For 19:2–12
Burban C, Petit RJ, Carcreff E, Jactel H (1999) Rangewide variation of the maritime pine bast scale Matsucoccus feytaudi Duc. (Homoptera: Matsucoccidae) in relation to the genetic structure of its host. Mol Ecol 8(10):1593–1602. https://doi.org/10.1046/j.1365-294x.1999.00739.x
Cheddadi R, Vendramin GG, Litt T, Francois L, Kageyama M, Lorentz S, Laurent JM, de Beaulieu JL, Sadori L, Jost A, Lunt D (2006) Imprints of glacial refugia in the modern genetic diversity of Pinus sylvestris. Glob Ecol Biogeogr 15(3):271–282. https://doi.org/10.1111/j.1466-822x.2006.00226.x
Dering M, Kosiński P, Wyka TP, Pers−Kamczyc E, Boratyński A, Boratyńska K, Reich PB, Romo A, Zadworny M, Żytkowiak R, Oleksyn J (2017) Tertiary remnants and Holocene colonizers: genetic structure and phylogeography of Scots pine reveal higher genetic diversity in young boreal than in relict Mediterranean populations and a dual colonization of Fennoscandia. Divers Distrib 23(5):540–555. https://doi.org/10.1111/ddi.12546
Devey ME, Bell JC, Smith DN, Neale DB, Moran GF (1996) A genetic linkage map for Pinus radiata based on RFLP, RAPD, and microsatellite markers. Theor Appl Genet 92(6):673–679. https://doi.org/10.1007/Bf00226088
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11(12):2571–2581. https://doi.org/10.1046/j.1365-294X.2002.01650.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinforma 1:47–50
Haesaerts P, Borziac I, Chekha VP, Chirica V, Drozdov NI, Koulakovska L, Orlova LA, van der Plicht J, Damblon F (2010) Charcoal and wood remains for radiocarbon dating Upper Pleistocene loess sequences in Eastern Europe and Central Siberia. Paleogeogr Paleoclimatol Paleoecol 291(1-2):106–127. https://doi.org/10.1016/j.palaeo.2010.03.034
Hantemirova EV, Heinze B, Knyazeva SG, Musaev AM, Lascoux M, Semerikov VL (2017) A new Eurasian phylogeographical paradigm? Limited contribution of southern populations to the recolonization of high latitude populations in Juniperus communis L. (Cupressaceae). J Biogeogr 44(2):271–282. https://doi.org/10.1111/jbi.12867
Jaramillo-Correa JP, Beaulieu J, Bousquet J (2004) Variation in mitochondrial DNA reveals multiple distant glacial refugia in black spruce (Picea mariana), a transcontinental North American conifer. Mol Ecol 13(9):2735–2747. https://doi.org/10.1111/j.1365-294X.2004.02258.x
Naydenov K, Senneville S, Beaulieu J, Tremblay F, Bousquet J (2007) Glacial vicariance in Eurasia: mitochondrial DNA evidence from scots pine for a complex heritage involving genetically distinct refugia at mid-northern latitudes and in Asia Minor. BMC Evol Biol 7(1):233. https://doi.org/10.1186/1471-2148-7-233
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York, p 512
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144(3):1237–1245
Pyhäjärvi T, Salmela MJ, Savolainen O (2008) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet Genomes 4(2):247–254. https://doi.org/10.1007/s11295-007-0105-1
Sannikov SN, Petrova IV (2012) Phylogenogeography and genotaxonomy of Pinus sylvestris L. populations. Russ J Ecol 43(4):273–280. https://doi.org/10.1134/S1067413612040145
Sannikov SN, Petrova IV, Dymshakova OS, Cherepanova OE (2014) Genetic and phenotypic differentiation of Calluna vulgaris (L.) Hull. in Pritobolie and Europe. Russ J Genet 50(9):925–933. https://doi.org/10.1134/s1022795414090129
Semerikov VL, Semerikova SA, Dymshakova OS, Zatsepina KG, Tarakanov VV, Tikhonova IV, Ekart AK, Vidyakin AI, Jamiyansuren S, Rogovtsev RV, Kalchenko LI (2014) Microsatellite loci polymorphism of chloroplast DNA of scots pine (Pinus sylvestris L.) in Asia and eastern Europe. Russ J Genet 50(6):577–585. https://doi.org/10.1134/S1022795414040127
Semerikov VL, Putintseva YA, Oreshkova NV, Semerikova SA, Krutovsky KV (2015) Development of new mitochondrial DNA markers in Scots pine (Pinus sylvestris L.) for population and phylogeographic studies. Russ J Genet 51(12):1199–1203. https://doi.org/10.1134/S1022795415120108
Sinclair WT, Morman JD, Ennos RA (1999) The postglacial history of Scots pine (Pinus sylvestris L.) in Western Europe: evidence from mitochondrial DNA variation. Mol Ecol 8(1):83–88. https://doi.org/10.1046/j.1365-294X.1999.00527.x
Soranzo N, Alia R, Provan J, Powell W (2000) Patterns of variation at a mitochondrial sequence-tagged-site locus provides new insights into the postglacial history of European Pinus sylvestris populations. Mol Ecol 9(9):1205–1211. https://doi.org/10.1046/j.1365-294x.2000.00994.x
Tsai Y-HE (2011) PhyloGeoViz: a web-based program that visualizes genetic data on maps. Mol Ecol Resour 11(3):557–561. https://doi.org/10.1111/j.1755-0998.2010.02964.x
Vidyakin AI, Semerikov VL, Polezhaeva MA, Dymshakova OS (2012) Spread of mitochondrial DNA haplotypes in population of scots pine (Pinus sylvestris L.) in northern European Russia. Russ J Genet 48(12):1267–1271. https://doi.org/10.1134/s1022795412120150
Volkova VS, Arkhipov SA, Babushkin AE, Kulkova IA, Guskov SA, Kuzmina OB, Levchuk LK, Mikhailova IV, Sukhorukova SS (2002) Stratigraphy of oil and gas basins of Siberia. Cenozoic of West Siberia. Publ. House of SB RAS, Department “GEO”, Novosibirsk 246 pp.
Willis KJ, van Andel TH (2004) Trees or no trees? The environments of central and eastern Europe during the last glaciation. Quat Sci Rev 23(23-24):2369–2387. https://doi.org/10.1016/j.quascirev.2004.06.002
Acknowledgements
The authors further acknowledge EG Phylippov, SN Sannikov, KG Zatsepina, S. Jamiyansuren, AK Ekart, LI Kalchenko, and RV Rogovtsev for the material collection and OS Dymshakova, NV Semerikov and MA Polezhaeva for the laboratory assistance. The authors thank two reviewers and the Associate Editor for useful comments, corrections, and suggestions that greatly helped us improve our manuscript.
Funding
This study was funded by the research grants No. 16-04-00607 from the Russian Basic Research Foundation and No. 14.Y26.31.0004 from the Government of the Russian Federation.
Author information
Authors and Affiliations
Contributions
VLS, VVT, AIV, and KVK collected the samples, generated and analyzed data, and wrote the manuscript. SAS and IVT established the experiment, isolated and genotyped DNA, and helped with the sample collection. YAP and NVO helped with marker development, sequence analysis, data interpretation, and manuscript editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Data archiving statement
A list of the population samples including mitotype frequency is provided in Supplementary material Table S1. Sequences used in the study were deposited in GeneBank (KY302807, KY302806, and KY302808).
Additional information
Communicated by S. C. González-Martínez
Electronic supplementary material
ESM 1
(DOCX 6888 kb)
Rights and permissions
About this article

Cite this article
Semerikov, V.L., Semerikova, S.A., Putintseva, Y.A. et al. Colonization history of Scots pine in Eastern Europe and North Asia based on mitochondrial DNA variation. Tree Genetics & Genomes 14, 8 (2018). https://doi.org/10.1007/s11295-017-1222-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1222-0