
Abstract
As an ecologically and economically important endemic bamboo species, moso (Phyllostachys edulis) has been widely distributed in Southern China. In the paper, 20 fluorescently labeled microsatellite markers were used to evaluate the genetic structure of Ph. edulis including 34 representative populations (803 individuals) covering the geographic range in China. Moderate genetic diversity (H = 0.376) and differentiation (Gst = 0.162) were detected at the species level, with the majority of genetic diversity occurring within populations (84.55%). Bayesian model-based structure analysis and sNMF/ANLS-AS method revealed the presence of two and three clusters. When K = 2, majority of populations (except SX) were clustered together (C1). It implied that SX, known as an introduced and isolated founder population, significantly differed from other populations for distinct environmental selection and allele mutation with the proof of scarce outcrossing and relatively high frequency of private allele. While K = 3, two subgroups (C1a of 18 populations and C1b of 14 populations) were detected within C1. The C1b displayed as a belt-shape region with an east-west direction. It coincided with the extremely high artificial selection in C1b (lower genetic diversity than that of C1a) due to the intensive plantation in the last four decades. Our results implied that the population protection and germplasm collection of moso bamboo should be not only from representative populations in Zhejiang, Jiangxi, Fujian, and other places with intensive cultivation in the east of China, but from populations with high level of gene and genotypic diversity in the west (e.g., HN5, GD1, GZ2, YN1, and SX).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Moso bamboo, Phyllostachys edulis (Carrière) J. Houz. (Poaceae), native to China, is the most economically and ecologically important bamboo species in China. As the third largest source of timber and the predominant source of bamboo shoots, it occupies approximately 70% of bamboo plantations and produces 5 billion US dollars annually in China (Fu 2001; Peng et al. 2010). Significantly commercial interest and robust clonality of this bamboo has resulted in wide cultivation in Southern China (Zhou 1998). However, greater disturbances by spontaneous vegetation as a result of human activity have led to habitat deterioration and some loss of germplasm resource. An accurate assessment of genetic diversity, population differentiation, and spatial structure of moso bamboo across its entire distribution range in China is crucial for the design and implementation of appropriate conservation strategies and utilization of biodiversity in natural and domesticated species.
In recent years, studies about the genetic diversity of Ph. edulis have been taken using different molecular markers. Lai and Hsiao (1997) revealed a very limited genetic diversity of Ph. edulis in Taiwan by random amplification of polymorphic DNA (RAPD) marker. Zhang et al. (2007) also studied 18 populations in mainland China by RAPD marker and exhibited a low genetic variation in Ph. edulis. Ruan et al. (2008) studied the genetic diversity and relationships of Ph. edulis from 17 provenances applying markers amplified-fragment length polymorphism (AFLP) and inter-simple sequence repeat (ISSR), which produced low polymorphism at species level (38 and 39%, respectively). Microsatellite markers, also known as simple sequence repeats (SSR), are particularly valuable in plant breeding programs because they are polymorphic, codominant, relatively abundant, and widely dispersed across the genome (Powell et al. 1996). With the rapid development of genomic SSR and EST-SSR sequences, microsatellite (SSR) markers have been developed for Ph. edulis and several other bamboo species in recent years and applied to estimate their transferability to other bamboo species, outcrossing rates, and genetic diversity in partial Ph. edulis populations and identify bamboo interspecies hybrids (Jiang et al. 2013; Kitamura and Kawahara 2009; Kitamura et al. 2009; Lin et al. 2014; Miyazaki et al. 2009; Tang et al. 2010). However, detailed insight into the population genetics of Ph. edulis across its entire distributions in China is still lacking as very few systematic studies have been conducted.
To identify levels and structure of genetic variation and initiate development of long-term strategies for sustainable use of resources, a widespread analysis of population variation in Ph. edulis in China was undertaken. In the present study, 20 fluorescently labeled polymorphic microsatellite markers were used to evaluate the genetic diversity and population structure of Ph. edulis including 34 representative populations (803 individuals) from across its geographic range in China. Our major concerns here are to reveal the current degree of genetic diversity, population differentiation, and spatial structure of moso bamboo in China and infer the main factors causing the population status. Specifically, our interests are (1) as a predominantly clonal reproduction species, whether the genetic diversity of moso bamboo is comparable with other sexual species. (2) What was the major force resulting to population differentiation and spatial structures? (3) As two known artificial plantations, where did the SX and SD populations originate from?
Materials and methods
Plant collections
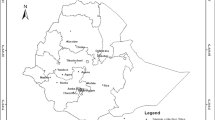
A total of 34 natural populations were sampled from almost all of the main growing regions of Ph. edulis in China. Within each population, leaves were collected randomly from each individual at least 50 m apart and then preserved in silica gel for further analysis. The geographic distribution, population code, collection sites, and sample sizes of the population are given in Fig. 1 and Table 1. All the 803 samples from 34 populations were analyzed in the research, and the vouchers for each population were deposited at the Bamboo Research Institute of Nanjing Forestry University, Nanjing, Jiangsu, China.

Geographic distribution of the 34 sampled populations of Ph. edulis. See Table 1 for population codes
DNA extraction
The genomic DNA was isolated from dried leaves following the slightly modified protocol of Doyle and Doyle (1991). Approximately 500 mg of silica-dried leaf tissue was crushed in liquid nitrogen for each sample, followed by further maceration in 2×CTAB buffer, 20 mM EDTA (pH 8), and 1.4 M NaCl and incubation at 65 °C for 1 h. DNA was separated from cellular debris by mixing with chloroform: isoamyl alcohol (24:1 v/v) followed by centrifugation at 13,000 rpm for 10 min, and the resulting aqueous layer was precipitated with 95% ethanol. The DNA pellet was air-dried and suspended in 50 μl 1×TE buffer at room temperature for 24 h.
Microsatellite genotyping
Twenty polymorphic microsatellite markers that were previously developed by our lab (Jiang et al. 2013) were selected to study the genetic structure of Ph. edulis (SSR references in Supplementary Table S1). The forward primer of each primer pair was labeled with one fluorescent dye (6-FAM or HEX). First, each of the 803 individuals were separately amplified by polymerase chain reaction (PCR) in a 10-μl reaction volume containing 30 ng of genomic DNA, 1 μl of 10×PCR buffer, 0.1 mM dNTPs, 0.87 mM MgCl2, 0.48 μM of each primer, and 0.5 U of Taq DNA polymerase (Takara Biotechnology Co., Shiga, Japan). All PCR reactions were performed in an Eppendorf Mastercycler gradient PCR thermal cycler (Hamburg, Germany) using a modified touchdown protocol (Don et al. 1991): 5-min denaturation at 94 °C, followed by 12 cycles of 94 °C for 30 s, 62 °C decreasing to 50 °C at 1 °C per cycle for 30 s, 72 °C for 30 s; 20 cycles of 94 °C for 30 s, 52 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. Finally, all PCR products were separated with GS500-ROX-labeled standard on an ABI 3730XL DNA Analyzer (Applied Biosystems Inc., Foster City, CA, USA), and the amplified fragment sizes were estimated with GenMapper 4.0 software (Applied Biosystems).
Data analysis
Overall genetic diversity for each population was assessed using mean number of alleles (Na), mean effective alleles (Ne), Shannon’s information index (I), percentage of polymorphic loci (Pp), average observed heterozygosity (Ho), average expected heterozygosity (He), and Wright’s fixation index (F). Genetic polymorphism for each locus was assessed by calculating polymorphism information content (PIC), inbreeding coefficient (Fis, Fit), and Nei (1973) genetic statistic, including mean diversity within each population (Hs), total genetic diversity in the pooled populations (Ht), total genetic diversity distributed among populations (Dst), and the proportion of genetic diversity that resides among populations (Gst = Dst/Ht). We also calculated the Wright’s F statistics (Fst) to illuminate the genetic differentiation of the 34 Ph. edulis populations and estimate the gene flow (Nm) among populations with the equation Nm = (1 − Fst)/4Fst (Wright 1978). The codominant data were performed using the GenAlEx 6.5 software to calculate Na, Ne, Pp, He, Ho, I, Fis, Fit, Fst, and Nm (Peakall and Smouse 2012b). The PIC value of each SSR marker and Nei’s pairwise genetic distance (Ds) were calculated using the PowerMarker 3.25 software (Liu and Muse 2005). Hs and Ht were calculated using PopGene 1.31 software (Yeh et al. 2000). Analysis of molecular variance (AMOVA) within populations and among populations was performed in Arlequin 3.5 software (Excoffier and Lischer 2010) to determine the distribution of variation at different hierarchical levels. The statistical significance of the variance components was tested with 1000 permutations. A Mantel test for a significant relationship between the genetic distances between sampling sites and their geographic distances was analyzed by the IBD program (Bohonak 2002).
Several genetic diversity parameters prefering clonal or partial clonal reproduction species were estimated for our data. These parameters reflect the genotypic richness (observed number of multilocus genotype (MLG) and expected MLG (eMLG)), diversity (Shannon-Wiener index (H), Stoddart and Taylor’s index (G), and Simpson’s index (lambda)), and evenness (E.5) of each population (Grunwald et al. 2003; Shannon 2001; Simpson 1949; Stoddart and Taylor 1988). A parameter named the index of association (I A ), which was useful to determine if populations are clonal or sexual, was also estimated (Brown et al. 1980). Nine hundred ninety-nine permutations were set for our data in order to give us a p value for I A . All parameters above were implemented by the poppr R package (Kamvar et al. 2014).
The Bayesian model-based program STRUCTURE version 2.3.4 (Pritchard et al. 2000) was used to delineate clusters of 803 individuals of the Ph. edulis populations on the basis of their genotypes at multiple loci. Two sets of runs were performed using the admixture model and correlated allele frequencies between populations. Initial runs were performed with 10,000 burn-in length (iteration) and 100,000 Markov Chain Monte Carlo (MCMC) replicated 15 times at each K from 1 to 34. The probable number of clusters was estimated by the likelihood of the probability of data L (K) (=LnP (D)). A second run was performed with 50,000 for burn-in length and 500,000 for MCMC replicates 15 times at each K. The best K value was estimated according to either L(K) or an ad hoc quantity ΔK (Evanno et al. 2005). After obtaining the optimum number of subpopulations, an AMOVA was performed using Arlequin 3.5 software (Excoffier and Lischer 2010) to estimate the genetic variance components within subpopulations and between subpopulations. Based on the microsatellite variation, MEGA 5.1 software (Tamura et al. 2011) was used to construct a Neighbor-Joining (NJ) tree, according to a pairwise population matrix of Nei’s pairwise genetic distances (Ds). Principal coordinates analysis (PCoA) of Ds was performed using GenALEx 6.5 software (Peakall and Smouse 2012).
Due to the dominant clonal reproduction of Ph. edulis which may give rise to a distinct population structure from other sexual reproduction plant species, we also performed individual ancestry coefficient estimation base on the sparse non-negative matrix factorization (sNMF) via alternating non-negativity-constrained least squares algorithm with active set (ANLS-AS) method (Frichot et al. 2014; Kim and Park 2007). The cross-entropy criterion estimating for clusters choosing (Alexander and Lange 2011) was performed on R package LEA (Francois 2016; Frichot and Francois 2015). The number of ancestral populations (K) was set to 1–34, and each K was repeated 100 times. The algorithms for estimating population structure implemented in LEA differ from those of structure dramatically, and the assessment can be more accurate than those of structure in the presence of inbreeding (Frichot et al. 2014). After the rational K was determined, we computed the spatial estimates of admixture coefficients and represented the spatial predictions on a geographic map (Francois 2016; Jay et al. 2012).
Results
Genetic diversity analysis
A total of 169 alleles were detected across 20 microsatellite loci with an average of 8.450 alleles per SSR locus for all 803 samples (see Table S2 in supplemental data). The mean observed number of alleles (Na) and expected number of alleles (Ne) of 34 populations were 2.287 and 1.657, respectively (Table 2). Fifteen populations (44%) presented private alleles. The percentage of polymorphic loci (Pp) with 34 populations varied from 50% (GX1) to 100% (GD1) with an average of 74.410%. Values for the mean expected heterozygosity (He) of the 34 populations ranged from 0.238 to 0.538, with an average of 0.320. Shannon’s information index (I) ranged from 0.329 to 0.948, with an average of 0.500. Based on 20 microsatellite markers, we detected moderate genetic diversity (H = 0.376) at the species level (see Table S2 in supplemental data). HN5 had the highest level of genetic diversity (He = 0.538; I = 0.948), followed by GD1 (He = 0.524; I = 0.936), while GX1 possessed the lowest genetic diversity (He = 0.238; I = 0.329). Those striking negative values of F (≤ -0.1) indicated heterozygote excess or homozygote deficiency at most loci in lots of populations (except GZ2, GD1, HN5, SX, SC2, GZ1, YN1, HN1, and HB2) (Table 2). It implied that those populations have been outcrossing more or less and undergone a high degree of heterozygote-oriented environmental or artificial selection pressures.
The genotype-based genetic diversity refers to genotypic richness (multilocus genotype (MLG), expected MLG), diversity (H, G, lambda), and evenness (E.5) of all 34 moso populations, which was estimated for its dominant asexual reproduction (Table 3). The arguments reflecting genotypic richness and diversity exhibited similar results among populations with allele-based genetic diversity analyses. The populations GD1 and HN5 displayed relatively high levels of genotypic richness (MLGGD1 = 18, eMLGGD1 = 14.976; MLGHN5 = 16, eMLGHN5 = 13.894) and diversity (H GD1 = 2.834, G GD1 = 16.030, lambdaGD1 = 0.938; H HN5 = 2.689, G HN5 = 13.444, lambdaHN5 = 0.926), while the GX1 population presented the lowest MLG (3), eMLG (2.696), H (0.456), G (1.292), and lambda (0.226), in all given populations. The genotypic evenness varied from population to population, and the populations with the highest and lowest E.5 were SC2 (0.427) and GD1 (0.939), respectively. Based on the estimation of the index of association (I A ), 21 of 34 populations (62%) presented asexual (or selfing) reproduction (pI A < 0.05). After clone correction, 13 of 34 populations (38%) still displayed non-sexual reproduction (pI A * < 0.05). This larger proportion of clonal populations partially supported the conventional view of the low frequency of sexual propagation in moso populations.
Based on the common multilocus genotype (MLG) across population analysis, we found only 26 of 265 (9.8%) MLGs occurred at two or more populations (Fig. 2), in which MLG.115 was the most common MLG and occurred at 14 populations, followed by MLG.259 (across 9 populations) and MLG.86 (across 8 populations). No MLG in populations GD2 and ZJ1 shared with other populations. Low proportion of common MLG means that private MLGs are frequently represented in each population. For instance, a total 17 of 18 MLGs in GD1, 13 of 16 MLGs in HN5, and 11 of 15 MLGs in GZ2 were unique across populations. Combining this result with the number of private alleles (Npa) in each population (Table 2), we inferred that a large part of private MLGs probably derived from distinct environmental selection pressure and mutations of alleles other than outcrossing.

The count of common multilocus genotypes (MLGs) across populations (a) and minimum spanning network based on Bruvo’s genetic distance for microsatellite markers (b). Nodes (circles) represent individual multilocus genotypes. Node colors/shades represent population membership proportional to the pie size. Node sizes are relatively scaled to log1.45n, where n is the number of samples in the nodes to avoid node overlap severely. Edges (lines) represent minimum genetic distance between individuals determined by Prim’s algorithm. Nodes that are more closely related will have darker and thicker edges whereas nodes more distantly related will have lighter and thinner edges
The minimal spanning network of MLG based on Bruvo’s genetic distance displayed that lots of MLGs have relatively close relationships except some private MLGs from SX, HN5, GD1, and GZ2 (Fig. 2b). The three most common MLGs (115, 86, and 259) were clustered in the center of the network, and the distance between MLG115 and 86 was closer. It corresponded to the result that MLG115 and 86 were more common in east populations, while MLG259 was common in west and central populations. Remarkably, HN5, GZ2, GD1, and SX populations may be multi-origin or mixed in the past due to their large genetic distances between some MLGs within each population (Fig. 2b).
Genetic structure analysis
As revealed by the estimated genetic diversity, the total diversity (mean Ht) and diversity within populations (mean Hs) were 0.376 and 0.320 across all populations, respectively (see Table S2 in supplemental data). Dst and Gst were 0.056 and 0.162 in the light of Ht and Hs. The results inferred that genetic variation in this species mainly existed within populations and genetic differentiation among populations was moderate. The F statistics results showed moderate genetic differentiation among 34 Ph. edulis populations (Fst = 0.172) and considerable gene flow within populations (Nm = 1.203).
Population structure of moso bamboo was analyzed using STRUCTURE 2.3.4 (Evanno et al. 2005). The distribution of Log-likelihood [LnP (D)] did not show a clear trend (Fig. 3a), and ΔK was then calculated to estimate the true value of K. The result of this calculation showed that when K = 2, ΔK reached its maximal value (Fig. 3b).

STRUCTURE analyses based on the microsatellite data of 803 individuals (34 populations) of Ph. edulis. a Mean Ln P(D) (± SD) over 15 runs for each K value. b The corresponding ΔK statistics calculated according to ΔK (Evanno et al. 2005). c Histogram of the structure analysis for the model with K = 2 and 3. The different colored bars (red, green, blue) refer to three different genetic pools (C1, C2, C3), and bar length is proportional to the inferred ancestry values into each group for each individual when K = 3. d Proportions of ancestry of each population in two (K = 2) and three (K = 3) gene pools defined by Pritchard et al. (2000)
As a result, these data showed that the population was defined as two subgroups. According to the inferred ancestry, most individuals showed an average estimated major membership proportion ≥ 0.60 (Supplementary Material Table S3, when K = 2); therefore, they could be classified as mainly belonging to one of the two distinct genetic groups according to their largest ancestry membership fraction. The majority of individuals from genetic cluster 1 (C1, Fig. 3c, when K = 2) comprised 32 populations (an average ancestry of 95%), and those from C2 mainly consisted of SX population (75.8%) (Fig. 3c, Table S3). Due to the proportion of membership lower than 0.6, HN5 was considered as a mixture of the two genetic clusters (Table S3, when K = 2). Therefore, K = 2 as the most likely number of populations seems overly conservative. More likely, K = 3 is a more meaningful value to obtain a clear genetic structure. Based on the proportion of ancestry of each population in the three clusters (Table S3, when K = 3), C1 contained most of the individuals from 19 populations (an average ancestry of 90%), C3 consisted of most of the individuals from 13 populations (an average ancestry of 82.2%), and C2 still mainly included the individuals from SX and HN5 populations (Fig. 3c when K = 3). It also showed clusters C1 and C3, when K = 3, were equivalent to subgroups (C1a and C1b) from C1 when K = 2. As revealed by Fig. 3d, the proportions of ancestry of each population in each of two and three gene pools were not well associated with the geographical areas.
Another population structure analysis using a different algorithm from STRUCTURE software by the R package LEA (Francois 2016) revealed similar results. The optimal number of ancestral populations (K) was two or three depending on the smaller cross-entropy criterion (Fig. 4a). When K = 2, almost all individuals from 33 populations (exclude SX) were clustered together (cluster1), except a few samples (mainly from HN5, GD1, GZ2) sharing high proportions of ancestry coefficients with a majority of samples from SX population (cluster2) (Fig. 4b, c). When K = 3, the primary cluster 1 while K = 2 was further divided into two clusters (the new cluster 1 and cluster 2), and the primary cluster 2 became a new cluster 3 (Fig. 4b, d). The new cluster 1 displayed as a belt-shape region with an east-west direction.

Ancestry coefficient analyses of 803 individuals from 34 populations of Ph. edulis based on the 20 microsatellite loci. a The cross-entropy criterion changed with the number of ancestral populations (K). Smaller values of the cross-entropy criterion usually mean better runs. b The bar plot of admixture coefficients among all 803 individuals; the order of individuals from left to right was same as Fig. 3c. c, d The spatial interpolation of ancestry coefficients based on the POPSutilities.R suite of functions (Jay et al. 2012). Only the cluster with the maximal local contribution to ancestry is represented at each geographic point of the map
Cluster analysis
A Neighbor-Joining tree was constructed based on Nei’s genetic distance (D S , Fig. 5a) for a better understanding of the relationships between these populations. Population SX was genetically distinct from other populations while two clusters were set. After setting the number of clusters as three, group 1 (populations with a blue tip label) included two populations (SX and HN5); group 2 included 18 populations mainly collected from south-western and central China, Guangdong and Fujian province; and group 3 included the rest 14 populations, which are located at south-eastern and central China. These cluster results were largely agreed with the conclusions from pop structure analyses. In addition, a NJ tree for 803 Ph. edulis samples was also constructed (Supplementary Fig. S1). The clustering pattern was found to be in general agreement with relationships based on ancestry studies, although several individuals from different geographic origins grouped together. Still, in most cases, the samples belonging to the same population are in the same branches. Isolation by distance analysis based on the Mantel test also indicated that genetic differences increase linearly with geographic distances (Supplementary Fig. S2).

Neighbor-Joining plot and principal coordinates analysis for 34 populations of Ph. edulis based on Nei’s genetic distances (D S ). a The tip labels are colored according to the structure simulation for K = 3. b Geographically distinct genetic clusters are indicated by squares. The first and second axes extracted 44.44 and 23.7% of the total genetic variation, respectively
The principal coordinates analysis (PCoA) result based on Nei’s genetic distances (D S ) reflected the genetic differentiation among the 34 Ph. edulis populations (Fig. 5b). The first three principal coordinates explained 78.4% of the total variation. PCo1 explained 44.44% of the total genetic variance, and PCo2 explained 23.7%. Based on the dispersion locations of different populations in the plot, large genetic divergence was observed in pairwise comparisons between SX, GZ2, HN5, and the other populations. The clustering of populations was in agreement with the Neighbor-Joining tree and STRUCTURE analysis.
AMOVA analysis
An AMOVA test was conducted according to the Bayesian-clustering results above (Table 4). All the 34 Ph. edulis populations were divided into cluster 1 (C1) and cluster 2 (C2). The C1 group was classified into two subgroups. The subgroups included C1a, containing 18 populations (SC1\2, GZ1, YN1\2, GD1\2, HN3\4, HB1\2, FJ1\2\3\4\5, JX3, and AH1) and C1b, comprised of 14 populations (GZ2, GX1\2, HN1\2, JX1\2\4, ZJ1\2\3, AH2, JS, and SD). The remaining populations (SX, HN5) belonged to C2. Wright (1978) proposed that there is moderate genetic differentiation when the Fst values are between 0.05 and 0.15 among populations. AMOVA results indicated that most of the existing genetic diversity was distributed within populations (88.55%) and only 15.45% occurred among populations (Fst = 0.154). Pairwise Fst showed a strong genetic differentiation (Fst = 0.253) between the two clusters. Moreover, high differentiation occurred between subgroups (C1a and C1b) within the C1 group (Fst = 0.175). Taken together, these results indicated moderate levels of genetic differentiation between populations of Ph. edulis.
Discussion
Genetic diversity
Levels of genetic diversity in moso bamboo have been measured in several studies. Lai and Hsiao (1997) observed a very limited genetic variation in this species from Taiwan based on 13 RAPD markers, three microsatellites, and one minisatellite marker. Ruan et al. (2008) observed the percentage of polymorphic loci (Pp) of 38 based on AFLP and Pp of 39.9% on ISSR markers from 17 Ph. edulis provenances. Zhang et al. (2007) observed a Shannon’s information index (I) value of 0.377 based on RAPD markers for 18 Ph. edulis populations. The within-population variation was detected for 12 Dendrocalamus membranaceus populations using 10 ISSR markers (Hs = 0.164, I = 0.249, and Pp = 48%) (Yang et al. 2012) and for 7 Dendrocalamus giganteus populations using 7 ISSR markers (Hs = 0.042, Pp = 11.33%) (Tian et al. 2012). In contrast, a moderate genetic diversity (Hs = 0.32, I = 0.499, Pp = 74.4%) was revealed within the 34 Ph. edulis populations analyzed herein. This distinction is likely due to the different sample size, sampling strategy, and markers used. Hamrick and Godt (1996) suggested that life form and breeding system had highly significant influences on genetic diversity and its distribution, and predominately outcrossing woody species had more genetic diversity than predominately herbaceous species. However, high genetic diversity has been observed in some clonal plants based on different molecular markers in recent years (Chen et al. 2010; Clark-Tapia et al. 2005; De Witte et al. 2012). Regardless of breeding system, the geographic distribution of populations, habitat characteristics, and population history have an important impact on the genetic diversity of clonal plants as well. Ph. edulis has a rather striking life history, characterized by a prolonged vegetative phase, long flowering intervals (67–120 years), and gregarious flowering (Isagi et al. 2016; Janzen 1976; Watanabe et al. 1994). Besides the low incidence of sexual reproduction and somatic mutations, genetic variation of Ph. edulis also may result from historical factors. As the most important economic bamboo species, Ph. edulis has a long history of cultivation, and artificial gene flow caused by migration may be one of the reasons explaining a rich genetic diversity observed in several populations.
In our study, the highest levels of genetic diversity and few sexual reproduction (pI A = 0.001), as well as high frequency of private alleles and MLGs, had been seen in populations HN5 (Huaihua, Hunan), GD1 (Renhua, Guangdong), and GZ2 (Liping, Guizhou) (Tables 2, 3, Fig. 2). Besides, the minimal spanning network displayed dispersed and relatively large distances between MLGs within each of these populations. Therefore, we inferred the rich genetic diversity may be a result of mixture from multi-populations in the past and they underwent lower level artificial selection. Meanwhile, we suggest that population SX (Zhouzhi, Shaanxi) also has a rich genetic variation explained by multi-origin. A cloned plant population may have been established by a number of multi-origin genotypes which now may be polyclonal and maintain a high genetic diversity (Chung et al. 2013). According to historical records and our fieldwork, moso bamboos from SX were cultivated and introduced from the Hunan and Hubei provinces from 60 years ago. In addition, some unknown origin of seedlings was mixed in, contributing to multiple sources of germplasm. Moreover, most of the MLGs in SX have minimal spanning network with those in HN5, which implied the founders mainly originated from HN5. No flowering behavior, so far, further demonstrated that the genetic diversity of the population was in agreement with the historic genotype diversity. We also noticed that most populations had low genetic variability, especially those from Zhejiang, Anhui, Fujian, and Jiangxi provinces. In these places, moso bamboo was affected by strong artificial selection (Zhou 1998), resulting in a sharp reduction of existing native bamboo area and genetic diversity.
Genetic differentiation and gene flow
AMOVA analysis showed that, on average, genetic variation within a population was larger than that among populations. This situation is common among outcrossing and vegetatively propagated perennial species which are generally highly heterozygous and maintain high levels of genetic variation within populations (Hamrick and Godt 1989). Previous work by Wright (1978) stated that the genetic differentiation among populations would be moderate when the value of Fst was among 0.05–0.25 and high when Fst was greater than 0.25. From the AMOVA analysis, the overall level of genetic diversity arising from the differentiation over all populations was moderate (Fst = 0.154, Gst = 0.162). This differentiation was generally lower than those reported in other woody bamboos, Dendrocalamus membranaceus (Gst = 0.252) (Yang et al. 2012) and D. giganteus (Gst = 0.847) (Tian et al. 2012), and similar to the result in barley (Fst = 0.18) (Rodriguez et al. 2012). The gene flow (Nm) was estimated with the value of Fst. The value of gene flow (Nm) less than 1.0 is generally regarded as the threshold value beyond which significant population differentiation occurs (Slatkin and Barton 1989). In the present study, the mean value of Nm was 1.203, indicating there was enough gene flow in this population to counteract genetic differentiation by gene drift (Hamrick et al. 1992). Because of the naturally long vegetative phase and low pollen dispersion caused by infrequent and spare flowering, as well as the differences in flowering times, seed dispersal is not the most probable explanation for gene exchange over a long distance (Zhang and Ma 1990). As an economically important bamboo species, Ph. edulis most likely has undergone human-mediated movement of genotypes among partial distribution areas.
Population structure
Genetic structure analysis both based on STRUCTURE and R package LEA indicated K = 2 as the most likely model for two clusters, the main cluster which most populations fell into (C1) and cluster two which only a few populations belonged to (C2). A strong genetic differentiation (Fst = 0.252) occurred between C1 and C2 according to AMOVA analysis. When K = 3, our result revealed that 32 populations from C1 were further divided into two subgroups (C1a with 18 populations and C1b with 14 populations). These clusters we defined are also supported by our genetic distance-based NJ and PCoA analysis (Fig. 5). Remarkably, because most of individuals from SX fell into C2 and some individuals from HN5, GZ2, and GD1 located in central China showed mixed ancestry of C1 and C2, we inferred that SX, as a known introduced population, probably originated from these populations above. The minimal spanning network of MLG based on Bruvo’s genetic distance (Fig. 2b) also supported the point.
According to historical records, bamboos were largely introduced and cultivated in a number of Northern China regions under the “South Bamboo Transfer to North” program of China around 60 to 70 years ago. We further verified the reliability of this historical record by clustering analysis in moso bamboo. According to historical records, the moso bamboo stands in Zhouzhi, Shaanxi (SX), Northwest China, mainly consisting of asexual clones from Hunan, Hubei, Jiangsu, or other provenances (Li 1995). Our data confirmed that the existing SX moso bamboo population mainly originated from Huaihua, Hunan provenance (HN5) (Figs. 2, 3, 4, 5), Central China. The moso bamboo forest of Laoshan, Shandong (SD), Northern China, could be established through different original clones from several eastern provenances such as Fujian. Zhejiang, and Jiangsu, during 1966 to 1972. Structure analysis in our study strongly supports that the existing SD moso bamboo populations shared the same cluster with most of the eastern populations. For instance, the SD population has the lowest genetic distance with FJ3 population (Shaxian, Fujian) and FJ2 population (Zhangping, Fujian) (Figs. 3, 4, 5). According to cultural records, moso bamboo was not present in Chishui, Guizhou (GZ1) until 200 years ago in the Qing Emperor Qianlong (1769 AD), and the existing GZ1 moso bamboo had been introduced and developed by Li Litai from Shanghang County, Fujian province. Our data also suggested that GZ1 population and SC1 population in Southwestern China and Fujian populations fell into the same sub-group, especially with a relatively close genetic distance with Wuyishan and Jian’ou, Fujian (FJ1 and FJ5).
Implications for the development of Ph. edulis
Due to habitat deterioration as a result of human activity, as well as a very weak capacity for reproduction and regeneration from seedlings, there is an urgent need for germplasm collection and conservation of Ph. edulis. The study herein revealed genetic variation for Ph. edulis occurs mainly within populations, so a protective strategy should focus on each population. Several populations with an abundant genetic variation, e.g., Huaihua of Hunan (HN5), Renhua of Guangdong (GD1), and Liping of Guizhou (GZ2), should be the focus of in situ conservation to some degree. Meanwhile, ex situ conservation should be carried out to sustain the maximum genetic diversity of this important bamboo in China. The ex situ germplasm resources might include any available seeds and vegetatively propagated materials taken from multiple populations representing different clusters. Conservation of the different germplasms is of utmost importance as each population derives from unique ancient clones and thus maintains significant agro-biodiversity.
Conclusions
Moderate genetic diversity and differentiation were revealed within the 34 Ph. edulis populations. Population structure analyses displayed the number of ancestral populations was two (C1 and C2) for 803 individuals from 34 populations. Except early introduced populations SX and multi-origin population HN5, Ph. edulis was further divided into two subclusters (C1a and C1b). C1b mainly contained 14 populations from the region undergoing intensive bamboo plantation (Zhejiang, Jiangsu, Anhui, Jiangxi, Hunan, and Guangxi), and C1a included the rest 18 populations mainly collected from southeastern (Fujian), south (Guangdong) and southwestern (Guizhou, Yunnan, and Sichuan) China. These results are useful for the germplasm conservation and efficient utilization of moso genetic resources.
References
Alexander DH, Lange K (2011) Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinf 12(1):246–246. https://doi.org/10.1186/1471-2105-12-246
Bohonak AJ (2002) IBD (Isolation By Distance): a program for analyses of isolation by distance. J Hered 93(2):153–154. https://doi.org/10.1093/jhered/93.2.153
Brown AHD, Feldman MW, Nevo E (1980) Multilocus structure of natural populations of HORDEUM SPONTANEUM. Genetics 96(2):523–536
Chen YY, Han QX, Cheng Y, Li ZZ, Li W (2010) Genetic variation and clonal diversity of the endangered aquatic fern Ceratopteris pteridoides as revealed by AFLP analysis. Biochem Syst Ecol 38(6):1129–1136. https://doi.org/10.1016/j.bse.2010.12.016
Chung MY, López-Pujol J, Chung MG (2013) Low genetic diversity in marginal populations of Bletilla striata (Orchidaceae) in southern Korea: insights into population history and implications for conservation. Biochem Syst Ecol 46:88–96. https://doi.org/10.1016/j.bse.2012.09.019
Clark-Tapia R, Alfonso-Corrado C, Eguiarte LE, Molina-Freaner F (2005) Clonal diversity and distribution in Stenocereus eruca (Cactaceae), a narrow endemic cactus of the Sonoran Desert. Am J Bot 92(2):272–278. https://doi.org/10.3732/ajb.92.2.272
De Witte LC, Armbruster GJ, Gielly L, Taberlet P, Stocklin J (2012) AFLP markers reveal high clonal diversity and extreme longevity in four key arctic-alpine species. Mol Ecol 21(5):1081–1097. https://doi.org/10.1111/j.1365-294X.2011.05326.x
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991) ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 19(14):4008. https://doi.org/10.1093/nar/19.14.4008
Doyle JJ, Doyle JL (1991) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Francois O (2016) Running structure-like population genetic analyses with R. R Tutorials in Population Genetics U Grenoble-Alpes:pages 1–9
Frichot E, Francois O (2015) LEA:AnRpackage for landscape and ecological association studies. Methods Ecol Evol 6(8):925–929. https://doi.org/10.1111/2041-210X.12382
Frichot E, Mathieu F, Trouillon T, Bouchard G, Francois O (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics 196(4):973–983. https://doi.org/10.1534/genetics.113.160572
Fu J (2001) Chinese moso bamboo: its importance. Bamboo 22:5–7
Grunwald NJ, Goodwin SB, Milgroom MG, Fry WE (2003) Analysis of genotypic diversity data for populations of microorganisms. Phytopathology 93(6):738–746. https://doi.org/10.1094/PHYTO.2003.93.6.738
Hamrick J, Godt M (1989) Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant population genetics, breeding and genetic resources. Sinauer Associates, Sunderland, pp 43–63
Hamrick J, Godt M (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond Ser B Biol Sci 351(1345):1291–1298. https://doi.org/10.1098/rstb.1996.0112
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. In: Population genetics of forest trees. New Forests 6:95–124
Isagi Y, Oda T, Fukushima K, Lian C, Yokogawa M, Kaneko S (2016) Predominance of a single clone of the most widely distributed bamboo species Phyllostachys edulis in East Asia. J Plant Res 129(1):21–27. https://doi.org/10.1007/s10265-015-0766-z
Janzen DH (1976) Why bamboos wait so long to flower. Annu Rev Ecol Syst 7(1):347–391. https://doi.org/10.1146/annurev.es.07.110176.002023
Jay F et al (2012) Forecasting changes in population genetic structure of alpine plants in response to global warming. Mol Ecol 21(10):2354–2368. https://doi.org/10.1111/j.1365-294X.2012.05541.x
Jiang WX, Zhang WJ, Ding YL (2013) Development of polymorphic microsatellite markers for Phyllostachys edulis (Poaceae), an important bamboo species in China. Appl Plant Sci 1(7). https://doi.org/10.3732/apps.1200012
Kamvar ZN, Tabima J, Grunwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kim H, Park H (2007) Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics 23(12):1495–1502. https://doi.org/10.1093/bioinformatics/btm134
Kitamura K, Kawahara T (2009) Clonal identification by microsatellite loci in sporadic flowering of a dwarf bamboo species, Sasa cernua. J Plant Res 122(3):299–304. https://doi.org/10.1007/s10265-009-0220-1
Kitamura K, Saitoh T, Matsuo A, Suyama Y (2009) Development of microsatellite markers for the dwarf bamboo species Sasa cernua and Sasa kurilensis (Poaceae) in northern Japan. Mol Ecol Resour 1470
Lai C, Hsiao J (1997) Genetic variation of Phyllostachys pubescens (Bambusoideae, Poaceae) in Taiwan based on DNA polymorphisms Botanical Bulletin of Academia Sinica 38
Li Y (1995) Bamboo garden with the great achievement of “South Bamboo Transfer to North” Shanxi. Forestry 06:29–30
Lin Y, Lu JJ, Wu MD, Zhou MB, Fang W, Ide Y, Tang DQ (2014) Identification, cross-taxon transferability and application of full-length cDNA SSR markers in Phyllostachys pubescens. Springer Plus 3:486
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2129. https://doi.org/10.1093/bioinformatics/bti282
Miyazaki Y, Ohnishi N, Hirayama K, Nagata J (2009) Development and characterization of polymorphic microsatellite DNA markers for Sasa senanensis (Poaceae: Bambuseae). Conserv Genet 10(3):585–587. https://doi.org/10.1007/s10592-008-9575-4
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci 70(12):3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28(19):2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Peng Z, Lu T, Li L, Liu X, Gao Z, Hu T, Yang X, Feng Q, Guan J, Weng Q, Fan D, Zhu C, Lu Y, Han B, Jiang Z (2010) Genome-wide characterization of the biggest grass, bamboo, based on 10,608 putative full-length cDNA sequences. BMC Plant Biol 10(1):116. https://doi.org/10.1186/1471-2229-10-116
Powell W, Machray GC, Provan J (1996) Polymorphism revealed by simple sequence repeats. Trends Plant Sci 1(7):215–222. https://doi.org/10.1016/S1360-1385(96)86898-0
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Rodriguez M, Rau D, O’Sullivan D, Brown AH, Papa R, Attene G (2012) Genetic structure and linkage disequilibrium in landrace populations of barley in Sardinia. Theor Appl Genet 125(1):171–184. https://doi.org/10.1007/s00122-012-1824-8
Ruan X, Lin X, Lou Y, Guo X, Fang W, Chen C (2008) Genetic diversity of Phyllostachys heterocycla var. pubescens provenances by AFLP and ISSR. J Zhejiang For Sci Technol 28:29–33
Shannon CE (2001) A mathematical theory of communication. ACM SIGMOBILE Mobile Computing and Communications Review 5:3–55
Simpson EH (1949) Measurement of diversity. Nature 163(4148):688–688. https://doi.org/10.1038/163688a0
Slatkin M, Barton NH (1989) A comparison of three indirect methods for estimating average levels of gene flow. Evolution 43(7):1349–1368. https://doi.org/10.1111/j.1558-5646.1989.tb02587.x
Stoddart JA, Taylor JF (1988) Genotypic diversity: estimation and prediction in samples. Genetics 118(4):705–711
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. https://doi.org/10.1093/molbev/msr121
Tang DQ, JJ L, Fang W, Zhang S, Zhou MB (2010) Development, characterization and utilization of GenBank microsatellite markers in Phyllostachys pubescens and related species. Mol Breed 25(2):299–311. https://doi.org/10.1007/s11032-009-9333-4
Tian B, Yang HQ, Wong KM, Liu AZ, Ruan ZY (2012) ISSR analysis shows low genetic diversity versus high genetic differentiation for giant bamboo, Dendrocalamus giganteus (Poaceae: Bambusoideae), in China populations. Genet Resour Crop Evol 59(5):901–908. https://doi.org/10.1007/s10722-011-9732-3
Watanabe M, Ito M, Kurita S (1994) Chloroplast DNA phylogeny of Asian bamboos (Bambusoideae, Poaceae) and its systematic implication. J Plant Res 107(3):253–261. https://doi.org/10.1007/BF02344252
Wright S (1978) Evolution and the genetics of populations: a treatise in four volumes. Vol. 4, Variability within and among natural populations. University of Chicago Press
Yang HQ, An MY, ZJ G, Tian B (2012) Genetic diversity and differentiation of Dendrocalamus membranaceus (Poaceae: Bambusoideae), a declining bamboo species in Yunnan, China, as based on inter-simple sequence repeat (ISSR) analysis. Int J Mol Sci 13(12):4446–4457. https://doi.org/10.3390/ijms13044446
Yeh F, Yang R, Boyle T, Ye Z, Xiyan J (2000) PopGene32, Microsoft Windows-based freeware for population genetic analysis, version 1.32
Zhang W, Ma N (1990) Vitality of bamboo pollens and natural pollination in bamboo plants. For Res 3:250–255
Zhang SF, Ma QX, Ding YL (2007) RAPD analysis for the genetic diversity of Phyllostachys edulis China forestry. Sci Technol 21:3
Zhou FC (1998) Cultivation and utilization of bamboo. Bamboo Res 1:11–20
Acknowledgements
This work was supported by the Special Fund for Forest Scientific Research in the Public Welfare from State Forestry Administration of China (no. 201504106). We wish to thank Xiaoting Shen, Pei Hu, Yi Fan, Jie Wang and Dr. Liang Chen for sampling assistance and Dr. Tao Xia for experimental suggestion. We thank the public and private owners for access to the sampling sites. The authors are also thankful to the anonymous reviewers whose comments and suggestions have greatly improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W.-W. Guo
Data archiving statement
All the 20 SSR primers used in this work are available in GenBank. The corresponding GenBank accession was provided in supplemental Table S1.
Rights and permissions
About this article

Cite this article
Jiang, W., Bai, T., Dai, H. et al. Microsatellite markers revealed moderate genetic diversity and population differentiation of moso bamboo (Phyllostachys edulis)—a primarily asexual reproduction species in China. Tree Genetics & Genomes 13, 130 (2017). https://doi.org/10.1007/s11295-017-1212-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1212-2