
Abstract
Eight pairs of chloroplast DNA (cpDNA) universal primers selected from 34 pairs were used to assess the genetic diversity of 132 pear accessions in Northern China. Among them, six amplified cpDNA fragments showed genetic diversity. A total of 24 variable sites, including 1 singleton variable site and 23 parsimony informative sites, as well as 21 insertion-deletion fragments, were obtained from the combined cpDNA sequences (5309–5535 bp). Two trnL-trnF-487 haplotypes, five trnL-trnF-413 haplotypes, five rbcL haplotypes, six trnS-psbC haplotypes, eight accD-psaI haplotypes and 12 rps16-trnQ haplotypes were identified among the individuals. Twenty-one haplotypes were identified based on the combined fragments. The values of nucleotide diversity (Pi), average number of nucleotide differences (k) and haplotype diversity (Hd) were 0.00070, 3.56408 and 0.7960, respectively. No statistical significance was detected in Tajima’s D test. Remarkably, the important cpDNA haplotypes and their representing accessions were identified clearly in this study. H_19 was considered as one of the ancient haplotypes and was a divergent centre. H_16 was the most common haplotype of the wild accessions. H_2 was the haplotype representing the most pear germplasm resources (46 cultivars and two wild Ussurian Pear accessions), followed by haplotype H_5 (30 cultivars, two wild Ussurian Pear accessions and four sand pears in outgroups) representing the cultivars ‘Dangshan Suli’ and ‘Yali’, which harbour the largest and the second largest cultivation areas in China. More importantly, this study demonstrated, for the first time, the supposed evolution routes of Pyrus based on cpDNA divergence in the background of pear phylogeny in Northern China.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to paleontological data, genus Pyrus L. of subfamily Pomoideae of family Rosaceae is believed to be of Tertiary or possibly even more ancient origin (Rubtsov 1944). It developed from 22 recognized primary species (Bell et al. 1996) into various species during a long history of cultivation by humans. In China, pear trees originated in the mountainous regions of southwestern China and spread westward and eastward. As one of the three most diverse cultivated pear centres (Vavilov 1951), China has more than 2000 pear germplasm resources safely preserved in the five national fruit germplasm repositories located in Liaoning, Jilin, Xinjiang, Hubei and Yunnan (Cao 2014). Among them, 13 species originated in China, including species with commercial cultivars such as Chinese White Pear (P. bretschneideri Rehd.), Chinese Sand Pear (P. pyrifolia (Burm.f.) Nakai), Sinkiang Pear (P. sinkiangensis Yü) and Ussurian Pear (P. ussuriensis Maxim.) (Pu and Wang 1963; Yu 1979). Many of these varieties have adapted to different environmental conditions and mature in different periods in China.
Provinces and cities in Northern China, including Beijing, Tianjin, Inner Mongolia, Xinjiang, Gansu, Ningxia, Shanxi, Shaanxi, Qinghai, Shandong, Henan, Hebei, Anhui as well as parts of Jiangsu, Liaoning, Jilin and Heilongjiang are rich in pear germplasm resources. These regions are suitable for cultivating the main cultivars of many pear varieties such as ‘Yali’, ‘Xuehuali’ and ‘Dangshan Suli’ of White Pear varieties and ‘Nanguoli’, ‘Jianbali’ and ‘Huagai’ of Ussurian Pear varieties. Moreover, many improved varieties such as ‘Cuiguan’, ‘Huangguan’ and ‘Zaosu’ as well as some varieties introduced from Japan, Korea and Europe are grown in these regions.
Molecular data have been widely applied in studies on genetic diversity and phylogeography of plant species to further understand their evolutionary processes (Montanari et al. 2013; Wuyun et al. 2013; Zong et al. 2014). As a complement to nuclear DNA and a maternally inherited biomarker, chloroplast DNA (cpDNA) has been proved to be a useful and powerful tool in population genetics and phytogeography (Liu 2006; Liu et al. 2012, 2013; Zong et al. 2014) because of its features of uniparental inheritance, nearly neutral evolution, low evolutionary rate and little or no recombination (Clegg and Zurawski 1992). In addition, unlike the nuclear genome, cpDNA can also be used to analyse genetic structure and evolutionary events (Petit et al. 1993; Katayama et al. 2012; Wuyun et al. 2013).
Research on genetic diversity in Pyrus has mainly focused on the identification and characterization of cultivars or species using different molecular markers such as random amplified polymorphic DNA (RAPD) (Teng et al. 2001, 2002), amplified fragment length polymorphism (AFLP) (Bao et al. 2008), restriction fragment length polymorphism (RFLP) (Iketani et al. 1998), simple sequence repeats (SSR) (Yamamoto et al. 2001, 2002a, 2002b; Bao et al. 2007; Cao et al. 2007; Katayama et al. 2007; Yao et al. 2010; Sehic et al. 2012; Urrestarazu et al. 2015) and non-coding regions of cpDNA (Kimura et al. 2003; Liu et al. 2012; Wuyun et al. 2013). There has been significant progress in studies on the cpDNA diversity of Chinese pears (Liu 2006; Liu et al. 2012, 2013; Wuyun et al. 2013). However, the genus Pyrus shows extremely low chloroplast genome diversity compared with other angiosperms (Katayama and Uematsu 2003). The conservative evolution of cpDNA is valuable for exploring the phylogenetic relationships at many taxonomic levels (Palmer et al. 1985). Despite chloroplast genome conservation, structural alterations such as inversions, translocations, deletions (gaps) and insertions found in hypervariable regions of cpDNA (for example, accD-psaI and rps16-trnQ regions) evolved at a faster rate than other regions. Moreover, these structural alterations could be used for phylogenetic analyses of Pyrus species (Liu et al. 2013; Katayama et al. 2012) and reconstructing plant phylogeny at higher taxonomic levels (Downie and Palmer 1992; Doyle et al. 1992; Katayama and Ogihara 1996). Specifically, two large deletions of the non-coding accD-psaI and rps16-trnQ regions in two hypervariable regions have been used to classify cpDNA into three important types: type A has no large deletions, type B contains a 229-bp deletion in the region of accD-psaI and type C possesses a 141-bp deletion in the region of rps16-trnQ. Katayama et al. (2012) identified 25 cpDNA haplotypes based on 36 mutations in the fragments of accD-psaI and rps16-trnQ from 21 Pyrus species originating from Asia, Europe and Africa, and they established a Median-joining network based on these 25 cpDNA haplotypes. Wuyun et al. (2013) identified 30 cpDNA haplotypes based on 32 mutations from the same two hypervariable regions of 186 wild pear accessions and generated a haplotype network to illustrate their genetic relationships. The two hypervariable regions containing two large deletions have been proven useful and applicable for the evaluation of genetic diversity or genetic relationships among accessions in Pyrus.
Reports on pear cpDNA diversity have focused on the local species in one province or area (Liu et al. 2012; Chang et al. 2014; Zong et al. 2014; Zhang et al. 2016). There have been few reports on cpDNA diversity of different Pyrus species in Northern China. Moreover, the phylogenetic relationships among the accessions in Northern China are not clear. Thus, the aim of the current research was to study the diversity of cpDNA of pear accessions in Northern China and explore the evolution routes of Pyrus based on cpDNA haplotypes.
Materials and methods
Plant materials
A total of 132 pear accessions were analysed in this study, including 31 Chinese P. ussuriensis cultivars, 12 P. ussuriensis wild accessions, 56 P. bretschneideri cultivars, 16 P. pyrifolia cultivars, nine P. sinkiangensis cultivars, two P. xerophila cultivars and six P. betuleafolia wild accessions. In addition, four P. pyrifolia cultivars (‘Choujuurou’ and ‘Housui’ both from Japan, ‘Jiangwan Tangli’ from Jiangxi and ‘Xiaomeili’ from Zhejiang), three P. communis cultivars (‘Bartlett’ and ‘Conference’ both from England and ‘Clapp’s Favourite’ from America) and two Malus domestica accessions (‘Ralls’ and ‘Malus baccata (L.) Borkh’ both from China) were used as outgroups (Table 1).
The 132 pear accessions originated from 16 provinces, including Xinjiang, Liaoning, Jilin, Heilongjiang, Henan, Hebei, Shandong, Qinghai, Gansu, Anhui, Yunnan, Guizhou, Sichuan, Jiangsu, Shanxi and Shaanxi (Fig. 2), and all were preserved in the Chinese National Pear Germplasm Repository in Research Institute of Pomology, Chinese Academy of Agricultural Sciences (Xingcheng, Liaoning), located from 40° 16′ N 120° 06′ E to 40° 50′ N 120° 50′ E. Young and healthy leaves of different accessions were collected from trees 10 m apart from each other in the spring of 2014 and maintained in silica gel until use.
DNA extraction and quality of determination
Genomic DNAs were extracted with a modified cetyl trimethyl ammonium bromide (CTAB) method as described by Doyle and Doyle (1987) and subjected to 1.2% agarose gel electrophoresis for quality examination. All good quality DNA samples were stored at −70 °C and adjusted to 10–30 ng μl−1 before use.
CpDNA universal primer pairs for PCR amplification
Thirty-four pairs of cpDNA universal primers (Supplementary Table S1) used to explore cpDNA diversity of pear accessions were previously reported (Taberlet et al. 1991; Demesure et al. 1995; Kelchner and Clark 1997; Dumolin-Lapegue et al. 1997; Small et al. 1998; Parducci and Szmidt 1999; Parani et al. 2000; Katayama et al. 2012) and synthesized by Sangon (Shanghai, China). Six random cultivars were selected for PCR amplification to select cpDNA universal primer pairs suitable for subsequent experiments. After PCR amplification, 5 μl of each PCR product was electrophoresed on a 2% agarose gel.
Amplification and sequencing of cpDNA fragments
PCR amplification was carried out in a 40 μl reaction containing 40 ng of DNA template, 0.4 μM of each primer, 200 μM of each dNTP (Tiangen, Beijing, China), 2 mM MgCl2 and 2 U rTaq DNA polymerase (Tiangen, Beijing, China) at the following cycling conditions: 90 °C for 3 min followed by 35 cycles of 90 °C for 30 s, 52 °C for 40 s and 72 °C for 90 s and a final extension at 72 °C for 10 min. PCR amplification conditions for primers cp11 were modified as 94 °C for 3 min followed by 35 cycles of 94 °C for 30 s, 56 °C for 40 s and 72 °C for 90 s and a final extension at 72 °C for 6 min.
PCR products were electrophoresed, purified from agarose gels and analysed directly by an ABI 3730 sequencer system (Applied Bio systems, Inc., USA). The amplified fragment size was calculated based on an internal DNA standard with Gene Mapper 4.0 software (Applied Bio systems, Inc., USA).
Chloroplast haplotype analyses
Chloroplast DNA regions were aligned using software Clustal X ver2.1 (http://www.clustal.org/download/current/) and then analysed by MEGA ver6.06 (http://www.me-gasoftware.net/index.php). All sequences were saved in both FASTA and MEGA formats for further analyses after being refined manually. Chloroplast DNA fragments from the regions of trnL-trnF, trnL-trnF, rbcL, trnS-psbC, accD-psaI and rps16-trnQ were combined by using software PAUP beta ver4.0 (Swofford 2002) (http://www.sciencesoftware.com .cn/search/search_soft_detail12.a-sp.?id=752) for further analysis.
Haplotype number (h), haplotype diversity (Hd), variance of haplotype diversity (Vh), standard deviation of haplotype diversity (Sh), nucleotide diversity (Pi) (Nei and Li 1979), average number of nucleotide differences (k), variable (polymorphic) sites (Vs), singleton variable sites (Ss), parsimony informative sites (Ps) and insertion-deletion fragments (Ig) were calculated based on each cpDNA region and combined regions by using software DnaSP ver5.10.01 (Librado and Rozas 2009).
Tajima’s test
Tajima’s D values were calculated by using software DnaSP ver5.10.01 (Librado and Rozas 2009). A positive Tajima’s D signifies low levels of both low and high frequency polymorphisms, indicating a decrease in balancing selection. A negative Tajima’s D signifies an excess of low frequency polymorphisms relative to expectation, indicating purifying selection (Tajima 1989).
Construction of a haplotype network
The median-joining network was constructed with software Network ver4.6.13 (http://www.fluxus-engineering.com/) based on the cpDNA haplotypes derived from combined regions.
In addition, a complementary approach for reconstruction of a phylogenetic tree based on the plastid data was performed using software TCS ver1.21 (Clement et al. 2000).
Results
Six universal primer pairs for PCR amplification of cpDNA regions
Electrophoresis analysis showed that six cpDNA universal primer pairs, P02 (trnL-trnF), P03 (trnL-trnF), P09 (trnS-psbC), cp03 (rbcL), cp11 (accD-psaI) and cp19 (rps16-trnQ) produced clear, stable and single bands with mutation sites existing in each region.
Genetic diversity and chloroplast haplotypes based on each region and the combined regions and Tajima’s test
The trnL-trnF intergenic region was amplified by primer pairs P02, P03, P14 and cp13, all of which produced clear, stable and single bands. However, mutations were only detected in the sequences amplified by the former two primers. The fragments amplified by primer pairs P02 and P03 were 487 and 403–413 bp, respectively. The rbcL and trnS-psbC cpDNA fragments were aligned into 1270 and 1517 bp, respectively. The lengths of accD-psaI and rps16-trnQ ranged from 725 to 982 bp and 719 to 930 bp, respectively. The length of the combined fragments ranged from 5309 to 5535 bp after alignment.
The polymorphic information based on cpDNA fragments was analysed and is depicted in Table 2. We found one singleton variable site, 23 parsimony variable sites and 21 insertion-deletion fragments in the combined cpDNA regions. Moreover, we observed three parsimony informative sites in trnL-trnF-487, four parsimony informative sites in trnL-trnF-413, five parsimony informative sites in rbcL, one singleton variable site and four parsimony variable sites in trnS-psbC, two parsimony variable sites in accD-psaI and one parsimony variable site in rps16-trnQ. Among the six cpDNA regions, trnL-trnF-413 had two insertion-deletion gaps with lengths of 8 and 10 bp (Table S2); accD-psaI had six insertion-deletion gaps with lengths of 1, 2, 5, 10, 22 and 229 bp (Table S4), and rps16-trnQ had 13 insertion-deletion gaps with lengths of 1, 1, 2, 2, 5, 7, 8, 11, 17, 23, 24, 24 and 141 bp (Table S5). No gaps were found in the rbcL and trnS-psbC regions (Table S3).
Five parsimony informative sites were found in the 229-bp insertion sequence of accD-psaI (Table S4) and two were found in the 141-bp insertion sequence of rps16-trnQ (Table S5). Moreover, the non-coding region trnL-trnF-413 showed the highest nucleotide diversity (Pi = 0.00233) and an average number of nucleotide differences (k = 0.91869) (Table 2), while the non-coding region rps16-trnQ showed the lowest nucleotide diversity (Pi = 0.00006) and an average number of nucleotide differences (k = 0.04476).
Two trnL-trnF-487 haplotypes, five trnL-trnF-413 haplotypes, five rbcL haplotypes, six trnS-psbC haplotypes, eight accD-psaI haplotypes and 12 rps16-trnQ haplotypes were identified among individuals (Table 3). As one of the hypervariable regions of pear cpDNA, intergenic region rps16-trnQ had the most haplotypes and the highest haplotype diversity (h = 12, Hd = 0.7739, Table 3), followed in turn by another hypervariable cpDNA region accD-psaI (h = 8, Hd = 0.7604, Table 3) and the intergenic region trnL-trnF-413 (Hd = 0.7061). The trnL-trnF-487 region had the fewest haplotypes and the lowest haplotype diversity (h = 2, Hd = 0.1411, Table 3). The trnS-psbC and rbcL regions showed the lowest (Vh = 0.00054, Sh = 0.023, Table 3) and the highest (Vh = 0.00174, Sh = 0.043, Table 3) variance and standard deviation of haplotype diversity, respectively.
As shown in Table 3, the Tajima’s D value was positive only in the trnL-trnF-413 and accD-psaI regions and showed no significant differences among the six cpDNA regions (P > 0.10).
CpDNA haplotypes characterized by mutations in six regions
Twenty-four haplotypes (21 haplotypes for pear accessions and three for outgroups) were identified among the individuals analysed in this study based on base change characters and gaps, of which two haplotypes, H_23 and H_24, belonged to Malus outgroup accessions and H_22 belonged to P. communis ‘Conference’ (Table 1). Haplotypes H_1 to H_22 were found in 2, 48, 12, 1, 36, 2, 2, 11, 3, 1, 1, 4, 2, 3, 1, 4, 1, 1, 1, 1, 1 and 1 pear accessions, respectively. Fig. 1 shows the type and number of haplotypes in each species. Five haplotypes were found in 12 wild P. ussuriensis accessions (66.7% in 18 wild pear accessions, 16.7% in H_2 and H_5, 8.3% in H_7, 33.3% in H_12, 25.0% in H_16), six wild P. betuleafolia accessions (33.3% in 18 wild pear accessions, 33.3% in H_8, 16.7% in H_16, H_17, H_18 and H_19) and in nine P. sinkiangensis cultivars (7.9% in 114 cultivars, 22.2% in H_1, H_ 6, H_9 and H_13, and 11.1% in H_2). Six haplotypes were detected in 31 P. ussuriensis cultivars (27.2% in 114 cultivars, 48.4% in H_2, 12.9% in H_5, 29.0% in H_8, 3.2% in H_7, H_9 and H_10). Seven haplotypes were identified in 56 P. bretschneieri cultivars (49.1% in 114 cultivars, 30.3% in H_2, 16.1% in H_3, 1.8% in H_4, H_11, H_20 and H_21, and 46.4% in H_5). Three haplotypes were detected in 16 P. pyrifolia cultivars (14.0% in 114 cultivars, 75.0% in H_2, 18.8 in H_3 and 6.2% in H_14). Two haplotypes were observed in two P. xerophila cultivars (1.8% in 114 cultivars, 50.0% in H_2 and H_15) and three P. communis cultivars (66.7% in H_14, and 33.3% in H_22). The four P. pyrifolia cultivars from China used as outgroup shared the same haplotype H_5.

Genetic relationships of 132 pear accessions and seven pear outgroups based on chloroplast DNA analyses. The accessions of each species are indicated using the same colour-code. Circle size is proportional to the number of individuals per haplotype. Each haplotype is labelled below the circle. Gaps are treated as the fifth state
Figure 1 also shows the species each haplotype contains. H_2 was composed of five pear species, including two wild P. ussuriensis accessions, 15 P. ussuriensis cultivars, 17 P. bretschneideri cultivars, 12 P. pyrifolia cultivars, one P. sinkiangensis cultivar and one P. xerophila cultivar and, therefore, was considered to be one of the main haplotypes in Chinese pear cultivars. H_5 was composed of three pear species, including two wild P. ussuriensis accessions, four P. ussuriensis cultivars, 26 P. bretschneideri cultivars and four P. pyrifolia cultivars. Importantly, cultivars ‘Dangshan Suli’ and ‘Yali’ from P. bretschneideri with the largest and second largest cultivation areas in China belonged to this haplotype.
Geographic distribution of cpDNA haplotype polymorphisms
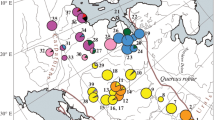
Twenty-one haplotypes were recognized from 132 pear accessions originating from 16 provinces (Table 1 and Fig. 2). Haplotypes H_1, H_4, H_6, H_10, H_11 and H_15 were only dispersed in pear accessions originating from Xinjiang, Shaanxi, Qinghai, Hebei, Liaoning and Gansu, respectively. The haplotypes H_17, H_18 and H_19 were only dispersed in P. betuleafolia originating from Shanxi. Haplotypes H_20 and H_21 were dispersed only in P. bretschneideri from Shandong. H_2, the most common haplotype, was dispersed in most pear germplasm resources from 15 provinces, except Henan.

The relative frequencies and geographic distributions of 21 haplotypes in 16 provinces of China. The 21 haplotypes H_1–H_21 are represented by 21 different colours and 21 numbers (1–21, see legend). AH Anhui, GS Gansu, GZ Guizhou, HB Hebei, HLJ Heilongjiang, HN Henan, JL Jilin, JS Jiangsu, LN Liaoning, QH Qinghai, ShX Shaanxi, SD Shandong, SX Shanxi, SC Sichuan, XJ Xinjiang, YN Yunnan
Haplotype network analysis
The median-joining network depicting the relationships among 24 cpDNA haplotypes (21 haplotypes for pear accessions and three for outgroups) derived from the comparison of the cpDNA sequences in the six regions was composed of six parts in different colours corresponding to circle C-1 through circle C-6 (Fig. 3). All haplotypes of pear accessions (H_1 to H_22) could be classified into three types: type A had no large deletion, type B had a 229-bp deletion in the region of accD-psaI and type C had a 141-bp deletion in the region of rps16-trnQ.

Median-joining network for cpDNA haplotypes in 132 pear accessions and outgroups based on six combined chloroplast DNA regions. The haplotypes are indicated by the yellow circles, the size of each circle being proportional to the observed frequency of each haplotype. Each circle, each node of each haplotype and the median vectors are labelled as C, H and mv, respectively
H_19 was unique to wild pear accession P. betuleafolia ‘Shanxi Duli 1’ and lay in the torso of the Median-joining network connecting directly or indirectly to the other haplotypes in 6 circles with different colours (Fig. 3). Circle C-2 contained four haplotypes including H_3, H_5, H_11 and H_21. All of them were type B haplotypes with a 229-bp deletion in accD-psaI and mainly represented Chinese White Pear cultivars. The circle C-3 contained six haplotypes. Among them, H_1, H_6, H_9 and H_13 were type C haplotypes with a 141-bp deletion in the rps16-trnQ region and represented the majority of Sinkiang Pear cultivars (eight out of nine, 88.9%), H_14 was a type C haplotype with a 141-bp deletion in the rps16-trnQ region and H_22 was a type A haplotype. Both H_14 and H_22 represented all the European pears (P. communis) used in this study.
Discussion
Relatively abundant cpDNA diversity and haplotype diversity of pear in Northern China
The accD-psaI and trnL-trnF intergenic spacers displayed the most polymorphic sites in the study of genetic characterization of pear varieties (Kimura et al. 2003). Consistently, in our study, the hypervariable region trnL-trnF-413 possessed the highest nucleotide diversity (Pi = 0.00233) and an average number of nucleotide differences (k = 0.91869). Moreover, the hypervariable region accD-psaI showed the third highest values for Pi (Pi = 0.00086) and k (k = 0.61531).
It is well known that cpDNA is a maternally inherited marker that undergoes little or no recombination and exhibits high levels of genetic variations, such as insertions, deletions, translocations and inversions (Clegg and Zurawski 1992; Petit et al. 1993). Some cpDNA regions are ideal fragments for phylogenetic research, hybrid cultivar identification and genetic diversity research. CpDNA is quite conservative, and the main mutation types are point mutations and indels. The genetic diversity of pear cpDNA was quite low (Katayama and Uematsu 2003; Katayama et al. 2012). In this study, the nucleotide diversity of 132 pear accessions from Northern China was 0.00070, lower than the Pi (Pi = 0.00105) of Callery pear accessions in Zhejiang (Liu et al. 2012). However, the haplotype (gene) diversity (Hd = 0.7960) was slightly higher than that of Callery pears (Hd = 0.719). This was probably because the haplotype number (h) corresponding to Hd of the 132 pear trees in the study was 21, much higher than that of 10 of P. calleryana, indicating a relatively abundant genetic diversity of pear trees in Northern China.
Sixteen haplotypes were found in 114 Chinese pear cultivars, slightly higher than that in previous studies (Liu et al. 2012; Wuyun et al. 2013). The cultivars of P. bretschneideri harboured seven haplotypes (h = 7), followed by P. ussuriensis (h = 6) and P. sinkiangensis (h = 5). Both wild P. ussuriensis and P. betuleafolia accessions had five haplotypes and shared H_16, which is less than those of wild Ussurian Pear accessions in a previous study (Wuyun et al. 2013). In summary, compared to wild pears, the Chinese pear cultivars in Northern China showed a wide range of genetic diversity and haplotypes in cpDNA, consistent with the results of Wuyun et al. (2013).
Important pear cpDNA haplotypes and their relationships revealed by the median-joining network
H_19 was unique to P. betuleafolia ‘Shanxi Duli 1’ and lay in the torso of the Median-joining network (Fig. 3). Therefore, it was considered to be one of the ancient haplotypes and a divergent centre. H_16 was the joint of haplotypes in circles C-3 and C-4. In addition, it was also the shared haplotype of wild P. ussuriensis and P. betuleafolia accessions. H_2 was found in most pear germplasm resources (46 cultivars and two wild Ussurian Pear accessions), followed by haplotype H_5 (30 cultivars, two wild Ussurian Pear accessions and four sand pears in outgroups) in the cultivars ‘Dangshan Su- li’ and ‘Yali’, which had the largest and second largest cultivation areas in China. Both H_2 and H_5 were ancient haplotypes of pear.
Nucleotide substitutions were also found between these haplotypes. There was one nucleotide substitution between H_4 and H_20, two between H_2 and H_20 and three between H_2 and H_4. In addition, a 2-bp indel was found between H_3 and H_5. Two singleton variable sites in cpDNA sequence were also identified among H_1, H_13 and H_14. Thus, we concluded that they had a close kinship with each other. Similarly, only one single nucleotide difference was found between H_6 and H_9. H_22 was the only haplotype that did not belong to Type C in circle C-3 and had a cpDNA sequence that was obviously different from other haplotypes determined through alignment; thus, it was spatially separated in the analyses using mv10. Therefore, it had a relatively distant relationship with the remaining haplotypes in this circle.
H_7, H_16 and H_18 in C-4 belonged to type A and had a close relationship to each other. H_7 differed from H_16 only by an A↔G transition and from H_18 by a 24 bp direct repeat region and gap (AAGAA ATAAG AATCA ACTTC TATA), in agreement with Katayama et al. (2012).
H_8 and H_15 connected to each other via H_12. H_8 represented the majority of Ussurian cultivars while H_12 represented all wild Ussurian accessions. The three haplotypes had a relatively close kinship and only varied by a 4-bp indel between H_8 and H_12 and an 18-bp indel between H_12 and H_15.
The haplotypes of occidental pears and most of the oriental pears lay in different and even opposite directions in the median-joining network, and they had obvious differences, indicating that they evolved independently and had a distant relationship (Zhang et al. 2016).
Genetic relationships of pear accessions with important haplotypes
Pear is generally considered as a complicated population without a high amount of gene flow among different species. However, our results of combined sequence analyses of six cpDNA regions showed clear genetic relationships between and within wild and cultivated accessions.
‘Korla pear’, a member of H_2 in this study, has been cultivated for more than 1300 years. As a famous variety in Xinjiang and perhaps in China, it has drawn wide attention among Chinese scholars. Some scholars believed that it is a hybrid species between occidental and white pears (Yang 1985, Zou et al. 1986, Teng et al. 2001) while others tended to attribute it to White Pear (He et al. 2011, Yang 2010). Ma et al. (2004a, b) and Shan et al. (2010) considered it as Sinkiang Pear based on their studies using ISSR and RAPD markers. We found that ‘Korla pear’ shared H_2 with White Pear ‘Donghuang’ from Xinjiang province and had haplotypes different from other Sinkiang Pear accessions, implying that White Pear participated in their evolution. In addition, considering that cpDNA is of matrilineal inheritance and reflects the matriarchal evolutionary history, we deduced that the female parent of ‘Korla pear’ was most likely to be P. bretschneideri. Apart from P. bretschneideri and P. sinkiangensis pears, the haplotype of P. pyrifolia, P. ussuriensis and P. xerophila pears was also H_2. Pu et al. (1985, 1986) conducted cytological studies and found that they all had triploid germplasm and displayed a similar genetic background.
Cultivars ‘Yanbian Dashan’ and ‘Pingguoli’ collected from Yanbian were identical in the combined cpDNA sequences, and both belonged to H_3. H_3 was composed of nine Chinese White Pear accessions and three Chinese Sand Pear cultivars and had a 229-bp indel fragment in cpDNA. ‘Pingguoli’ is one of the excellent pear varieties and has a cultivation history in China of over 90 years. Although it has been reported that ‘Pingguoli’ was introduced from Gyeonggido of North Korea (Wu 1984), where the major pear cultivars were anti-cold Japanese pear accessions, its origin is still debated. Based on the Yanbian Fruit Tree Survey Section (in Jilin province) in 1952, ‘Pingguoli’ was originally introduced to China in 1921 and named ‘Lipingguo’ at that time. After years of breeding and cultivation, it was renamed as ‘Pingguoli’ (Jing 1989; Qu et al. 2002, 2003; Yang et al. 2010). Moreover, classification of ‘Pingguoli’ is also controversial. Some scholars believed that ‘Pingguoli’ belonged to P. bretschneideri (Qu et al. 2001, 2002, 2003; Ma and Zhang 2009; Lu and Zhang 2009; Yang et al. 2010), while others considered it as P. pyrifolia (Challice and Westwood 1973) or a hybrid (Wang 1988; Qu et al. 1990; Ma et al. 2004a). Our results showed that it was clustered and shared the same combined cpDNA sequences with the cultivar ‘Yanbian Dashan’ from Yanbian, indicating that it had a similar genetic background with P. pyrifolia.
Two Japanese sand pears and 34 Chinese pears from nine provinces, including cultivars of P. ussuriensis, P. bretschneideri and P. pyrifolia formed H_5 and possessed type B haplotype with a 229-bp indel fragment, exhibiting a closely related relationship. Our results are consistent with previous reports showing that Japanese sand pear cultivars and Chinese sand pear cultivars shared similar genetic backgrounds and exhibited a high degree of kinship (Teng et al. 2002; Shen et al. 2006; Lu et al. 2011). In this study, Ussurian pear cultivars ‘Xiaoxiangshui’ and ‘Yanbian Longjing’ from Northeastern China had the same cpDNA sequences and belonged to H_8 together with the cultivar ‘Yanbian Xiehuatian’ from Yanbian of Jilin. The results are consistent with the results of Cao et al. (2012), showing that ‘Xiaoxiangshui’ and ‘Yanbian Longjing’ shared the same SSR alleles and had a relatively close relationship with ‘Yanbian Xiehuatian’. Together, these results demonstrated that the above conclusion was reliable at both levels of nuclear and cpDNA genomes.
Exploration of the supposed evolution routes of Pyrus from the median-joining network
The haplotype of wild P. betuleafolia accession ‘Shanxi Duli 1’ from Qinyuan, Shanxi province, one of the pear divergent centres, was H_19 and located in the torso of the Median-joining network (Fig. 3). Therefore, its site of origin, Shanxi province was regarded as the starting point of the evolution routes (Fig. 4). In addition, H_19 belonged to type A, which was one of the three cpDNA types and assumed to be the most primitive cpDNA type in a previous study (Katayama et al. 2012), implying that selecting Shanxi province as the starting point of the evolution routes was feasible.

The map of evolution routes of Pyrus in Northern China. The arrow shows the evolution direction. AH Anhui, GS Gansu, GZ Guizhou, HB Hebei, HLJ Heilongjiang, HN Henan, JL Jilin, JS Jiangsu, LN Liaoning, QH Qinghai, ShX Shaanxi, SD Shandong, SX Shanxi, SC Sichuan, XJ Xinjiang, YN Yunnan. The colour of each haplotype corresponds to that in Fig. 2
H_16 represented the haplotype of three wild P. ussuriensis accessions and one wild P. betuleafolia accession from Gansu and Shanxi. There are several possible geographical evolutionary routes such as the route Gansu → Qinghai → Shanxi based on the analysis of circle C-3 and Gansu → Xinjiang or Gansu → Qinghai based on the analysis of circle C-4. Remarkably, Gansu was considered as a vital location, especially the famous Hexi Corridor. To test which putative route was more supported, two scenarios of population divergence (Fig. 5) were constructed and evaluated based on approximate Bayesian computation (ABC) using DIYABC ver2.0 (Cornuet et al. 2014). Individuals from Shanxi, Gansu, Qinghai and Xinjiang provinces were treated as Pop 1, Pop 2, Pop 3 and Pop 4, respectively. Logistic regression computation and direct estimate were used to calculate posterior probabilities for the two scenarios (Fig. 6). Both approaches are congruent and show maximum support for the first scenario, indicating that Shanxi → Gansu → Qinghai was more likely to be the evolution route.

Two biogeography scenarios constructed based on approximate Bayesian computation (ABC) using DIYABC. Pop 1, Pop 2, Pop 3 and Pop 4 mean all individuals collected from Shanxi, Gansu, Qinghai and Xinjiang, respectively. Scenario 1 means the route Shanxi → Gansu → Qinghai and scenario 2 means Shanxi → Gansu → Xinjiang. The time is set as t3 > t2 > t1

Logistic regression analysis (left) and direct estimate (right) of posterior probabilities for scenario 1 and scenario 2
Haplotypes of the occidental pear were H_14 and H_22, the latter being divergent in the Median-joining network (Fig. 3). H_1 and H_6 were the haplotypes of Xinjiang pears from Xinjiang and Qinghai, respectively. Both H_9 and H_13 were the haplotypes of pears mainly from Gansu and H_13 was also a part of Xinjiang. As shown in the circle C-3 of the Median-joining network, there was a relatively close relationship between Sinkiang pear and occidental pear, consistent with another research (Liu 2006). This relationship could be further explained in the right part of Fig. 1, showing that H_14 and H_22 belonging to occidental pears merged earlier than H_1 and H_13 belonging to Xinjiang pears. Therefore, we concluded that occidental pears participated in the evolution of Xinjiang pears via geographic evolutional route of Areas Abroad → Xinjiang. Whether pear from Xinjiang also spread to foreign areas was beyond the scope of our study. Moreover, the relationship between oriental pears and occidental pears needs to be further analysed using more materials.
Another route was concluded based on the haplotype information in circle C-6. H_12 belonged to all the wild P. ussuriensis accessions from Jilin and Heilongjiang, whereas H_8 mainly consisted of P. ussuriensis accessions from Hebei, Jilin and Liaoning, including two wild P. betuleafolia accessions from Hebei and Shanxi. The difference between wild and cultivated Ussurian Pear accessions in Northern China was a 4-bp indel (AAAA), showing a very close relationship. Moreover, H_12 and H_8 differed from H_19 by a 10-bp indel and a 1-bp indel, respectively. These indels may be the critical force of evolution. Our results support the theory that pear trees spread from Yanshan Mountain in Hebei to Liaoning, Jilin and Heilongjiang.
In summary, to the best of our knowledge, this is the first report exploring the evolution routes of Pyrus based on cpDNA divergence in the background of pear phylogeny in Northern China.
References
Bao L, Chen K, Zhang D, Cao Y, Yamamoto T, Teng Y (2007) Genetic diversity and similarity of pear cultivars native to East Asia revealed by SSR (simple sequence repeat) markers. Genet Resour Crop Evol 54:959–971
Bao L, Chen K, Zhang D, Li X, Teng Y (2008) An assessment of genetic variability and relationships within Asian pears based on AFLP (amplified fragment length polymorphism) markers. Sci Hortic 116:374–380
Bell R, Quamme H, Layne R, Skirvin R (1996) Pears. In: Janick J, Moore JN (eds) Fruit breeding, vol 1: tree and tropical fruits. Wiley, New York, pp 441–514
Cao Y (2014) Pear varieties in China. China Agriculture Press, Beijing (in Chinese)
Cao Y, Liu F, Gao Y, Jiang L, Wang K, Ma Z, Zhang K (2007) SSR analysis of genetic diversity of pear cultivars. Acta Horticulturae Sinica 34:305–310 (in Chinese)
Cao Y, Tian L, Gao Y, Liu F (2012) Genetic diversity of cultivated and wild Ussurian Pear (Pyrus ussuriensis Maxim.) in China evaluated with M13-tailed SSR markers. Genet Resour Crop Evol 59:9–17
Challice J, Westwood M (1973) Numerical taxonomic studies of the genus Pyrus using both chemical and botanical characters. Bot J Linn Soc 67(2):121–148
Chang Y, Cao Y, Zhang J, Tian L, Dong X, Zhang Y, Qi D (2014) Studies on genetic diversity of pear germplasm resources in Liaoning province of China based on chloroplast DNA analysis. Acta Horticulturae Sinica 41(7):1307–1316 (in Chinese)
Clegg M, Zurawski G (1992) Chloroplast DNA and the study of plant phylogeny: present status and future prospects. In: Soltis PS, Soltis DE, Doyle JJ (eds) Molecular systematics of plants. Chapman and Hall, New York, pp 1–13
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cornuet J, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin J, Estoup A (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30(8):1187–1189
Demesure B, Sodzi N, Petit R (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–134
Downie S, Palmer J (1992) Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In: Soltis PS, Soltis DE, Doyle JJ (eds) Molecular systematics of plants. Chapman and Hall, New York, pp 14–35
Doyle J, Doyle J (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19:11–15
Doyle J, Davis J, Soreng R, Garvin D, Anderson M (1992) Chloroplast inversion and the origin of the grass family (Poaceae). Proc Natl Acad Sci U S A 89:7722–7726
Dumolin-Lapegue S, Pemonge M, Petit R (1997) An enlarged set of consensus primers for the study of organelle DNA in plants. Mol Ecol 6:393–397
He Z, Wu B, Yang Q (2011) Identification of Pyrus sinkiangensis and classification discussion of ‘Korla pear’. Shanxi Fruits 5:36–39 (in Chinese)
Iketani H, Manabe T, Matsuta N, Akihama T, Hayashi Y (1998) Incongruence between RFLPs of chloroplast DNA and morphological classification in east Asian pear (Pyrus spp). Genet Resour Crop Evol 45:533–539
Jing Z (1989) The origin and development of Pingguoli. Northern Horticulture 1:21–22 (in Chinese)
Katayama H, Ogihara Y (1996) Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA. Curr Genet 20:527–581
Katayama H, Uematsu C (2003) Comparative analysis of chloroplast DNA in Pyrus species: physical map and gene localization. Theor Appl Genet 106:303–310
Katayama H, Adachi S, Yamamoto T, Uematsu C (2007) A wide range of genetic diversity in pear (Pyrus ussuriensis var. aromatica) genetic resources from Iwate, Japan revealed by SSR and chloroplast DNA markers. Genet Resour Crop Evol 54:1573–1585
Katayama H, Tachibana M, Iketani H, Zhang S, Uematsu C (2012) Phylogenetic utility of structural alterations found in the chloroplast genome of pear: hypervariable regions in a highly conserved genome. Tree Genet Genomes 8:313–326
Kelchner S, Clark L (1997) Molecular evolution and phylogenetic utility of the chloroplast rpII6 intron in Chusquea and the Bambusoideae (Poaceae). Mol Phylogenet Evol 8:385–397
Kimura T, Iketani H, Kotobuki K, Matsuta N, Ban Y, Hayashi T, Yamamoto T (2003) Genetic characterization of pear varieties revealed by chloroplast DNA sequences. J Hortic Sci Biotechnol 78:241–247
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liu Y (2006) Research on chloroplast DNA diversity of genus pear in China. Master Dissertation. Capital Normal University, Beijing. (in Chinese)
Liu J, Zheng X, Potter D, Hu C, Teng Y (2012) Genetic diversity and population structure of Pyrus calleryana (Rosaceae) in Zhejiang Province, China. Biochem Syst Ecol 45:69–78
Liu J, Sun P, Zheng X, Potter D, Li K, Hu C, Teng Y (2013) Genetic structure and phylogeography of Pyrus pashia L. (Rosaceae) in Yunnan Province, China, revealed by chloroplast DNA analyses. Tree Genet Genomes 9(2):433–441
Lu F, Zhang Y (2009) Taxonomic status identification of Ping guo pear by AFLP molecular marker. J Anhui Agric Sci 37:1937–1938 (in Chinese)
Lu J, Wu J, Zhang S, Wu H, Zhang Y (2011) Genetic diversity and polygentic relationship among pears revealed by SSR analysis. J Nanjing Agric Univ 34(2):38–46 (in Chinese)
Ma Y, Zhang Y (2009) The application of RAPD in status identification of ‘Pingguoli’. Chin Agric Sci Bull 25:71–73 (in Chinese)
Ma B, Niu J, Pan L, Feng J, Lu X (2004a) Study on the genetic relationships among 18 pear cultivars by RAPD. J Fruit Sci 21(6):521–525 (in Chinese)
Ma B, Niu J, Wu Z, Tan W (2004b) Molecular markers analysis of main cultivars relationship between Pyrus in Xinjiang. J Shihezi Univ (Nat Sci) 22(2):97–102 (in Chinese)
Montanari S, Saeed M, Knäbel M, Kim Y, Troggio M, Malnoy M et al (2013) Identification of Pyrus single nucleotide polymorphisms (SNPs) and evaluation for genetic mapping in European pear and interspecific Pyrus hybrids. PLoS One 8(10):e77022
Nei M, Li W (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A 76:5269–5273
Palmer J, Jorgensen R, Thompson W (1985) Chloroplast DNA variation and evolution in Pisum; patterns of change and phylogenetic analysis. Genetics 109:195–213
Parani M, Lakshmi M, Ziegenhagen B, Fladung M, Senthilkumar P, Parida A (2000) Molecular phylogeny of mangroves VII PCR-RFLP of trnS-psbC and rbcL gene regions in 24 mangrove and mangrove-associate species. Theor Appl Genet 100:454–460
Parducci L, Szmidt A (1999) PCR-RFLP analysis of cpDNA in the genus Abies. Theor Appl Genet 98:802–808
Petit R, Kremar A, Wagner D (1993) Geographic structure of chloroplast DNA polymorphisms in European oaks. Theor Appl Genet 87:122–128
Pu F, Wang Y (1963) Pomology of China. Pears, vol 3. Shanghai Science and Technology Press, Shanghai (in Chinese)
Pu F, Lin S, Song W, Chen R, Li X (1985) Research on the karyotypes of Pyrus in China I. Plant Sci J 3(4):381–387 (in Chinese)
Pu F, Lin S, Chen R, Song W, Li X (1986) Research on the karyotypes of Pyrus in China II. Acta Horticulturae Sinica 13(2):87–90 (in Chinese)
Qu Z, Pan J, Shan Z (1990) The log of Beijing fruit tree. Beijing Press, Beijing, pp 239–240 (in Chinese)
Qu B, Jin X, Chen Y, Liu H, Wang P (2001) RAPD analysis of germplasm resources in Pyrus. Acta Horticulturae Sinica 28:460–462 (in Chinese)
Qu B, Jin X, Chen Y, Liu H, Wang P (2002) Classification study of Pingguoli in Yanbian area using RAPD markers. Progress Hortic 5:178–182 (in Chinese)
Qu B, Jin X, Chen Y, Liu H, Wang P, Zheng D (2003) Classification study of Pingguoli in Yanbian area by using RAPD markers. J Jilin Agric Univ 25:292–295 (in Chinese)
Rubtsov G (1944) Geographical distribution of the genus Pyrus and trends and factors in its evolution. Am Nat 78:358–366
Sehic J, Garkava-Gustavsson L, Fernández-Fernández F, Nybom H (2012) Genetic diversity in a collection of European pear (Pyrus communis) cultivars determined with SSR markers chosen by ECPGR. Sci Hortic 145:39–45
Shan J, Li J, Luo S, Cheng Q, Li C (2010) Analysis of genetic relationships of pear germplasm resources in Xinjiang based on ISSR and RAPD. Xinjiang Agric Sci 47(9):1714–1721 (in Chinese)
Shen Y, Teng Y, Tanabe K (2006) RAPD analysis for genetic assessment of some cultivars of Pyrus pyrifolia derived from China and Japan. Acta Horticulturae Sinica 33(3):621–624 (in Chinese)
Small R, Ryburn J, Cronn R, Seelanan T, Wendel J (1998) The tortoise and the hare: choosing between non-coding plastome and nuclear ADH sequences for phylogeny reconstruction in recently diverged plant group. Am J Bot 85:1301–1345
Swofford D (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sinauer Associates, Sunderland MA. (Program)
Taberlet P, Gielly L, Pauton G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Tajima F (1989) Statistical method for testing the neutral mutation hypotheses by DNA polymorphism. Genetics 123:585–595
Teng Y, Tanabe K, Tamura F, Itai A (2001) Genetic relationships of pear cultivars in Xinjiang, China as measured by RAPD markers. J Hortic Sci Biotechnol 76:771–779
Teng Y, Tanabe K, Tamura F, Itai A (2002) Genetic relationships of Pyrus species and cultivars native to East Asia revealed by randomly amplified polymorphic DNA markers. J Am Soc Hortic Sci 127:262–270
Urrestarazu J, Royo J, Santesteban L, Miranda C (2015) Evaluating the influence of the microsatellite markerset on the genetic structure inferred in Pyrus communis L. PLoS One 10(9):e0138417
Vavilov N (1951) The origin, variation, immunity and breeding of cultivated plants. Chronica Botanica 13:1–366
Wang Y (1988) Deciduous fruit tree breeding. Press of Agriculture, Beijing, pp 192–193 (in Chinese)
Wu G (1984) The taxonomy of temperate-zone fruit trees in China. Press of Agriculture, Beijing, pp 33–80 (in Chinese)
Wuyun T, Ma T, Uematsu C, Katayama H (2013) A phylogenetic network of wild Ussurian pears (Pyrus ussuriensis Maxim.) in China revealed by hypervariable regions of chloroplast DNA. Tree Genet Genomes 9:167–177
Yamamoto T, Kimura T, Sawamura Y, Kotobuki K, Ban Y, Hayashi T, Matsuta N (2001) SSRs isolated from apple can identify polymorphism and genetic diversity in pear. Theor Appl Genet 102:865–887
Yamamoto T, Kimura T, Sawamura Y, Manabe T, Kotobuki K, Hayashi T, Ban Y, Matsuta N (2002a) Simple sequence repeats for genetic analysis in pear. Euphytica 124:129–137
Yamamoto T, Kimura T, Sawamura Y, Manabe T, Kotobuki K, Hayashi T, Ban Y, Matsuta N (2002b) Genetic linkage maps constructed by using an interspecific cross between Japanese and European pears. Theor Appl Genet 106:9–18
Yang H (1985) The application of palynology in the taxonomy of some plants in Pyrus. Fruit Sci 3:2–9 (in Chinese)
Yang C (2010) Genetic diversity of cytoplasm heredity in some main cultivars of Pyrus. Master Dissertation. Shihezi University, Xinjiang. (in Chinese)
Yang L, Li W, Qu B (2010) The classification discussion of ‘Pingguoli’. J Agric Sci Yanbian Univ 32:73–76 (in Chinese)
Yao L, Zheng X, Cai D, Gao Y, Wang K, Cao Y, Teng Y (2010) Exploitation of Malus EST-NSSRs and the utility in evaluation of genetic diversity in Malus and Pyrus. Genet Resour Crop Evol 57:841–851
Yu D (1979) Taxonomy of the fruit tree in China. Agriculture Press, Beijing (in Chinese)
Zhang J, Chen Q, Cao Y, Yang X, Fan J, Hu H (2016) Genetic diversty and phylogenetics of pear (Pyrus L.) germplasm resources from Hubei province revealed by chloroplast DNA variation. J Genet Resour 17(4):766–772 (in Chinese)
Zong Y, Sun P, Liu J, Yue X, Niu Q, Teng Y (2014) Chloroplast DNA-based genetic diversity and phylogeography of Pyrus betulaefolia (Rosaceae) in Northern China. Tree Genet Genomes 10:739–749
Zou L, Zhang X, Zhang Z, Song B, Guo S (1986) Research on genetic relationships of Pyrus based on pollen morphology. Acta Horticulturae Sinica 13(4):219–224 (in Chinese)
Acknowledgements
The study was sponsored by the National Natural Science Foundation of China (grant No. 31272128).
Data archiving statement
The authors declare that all the work described in this manuscript followed the standard Tree Genetics and Genomes policy. The sequences will be uploaded soon, and all the sequences in this study will be found in the National Center of Biotechnology Information (NCBI) database.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Chagné
Rights and permissions
About this article

Cite this article
Chang, YJ., Cao, YF., Zhang, JM. et al. Study on chloroplast DNA diversity of cultivated and wild pears (Pyrus L.) in Northern China. Tree Genetics & Genomes 13, 44 (2017). https://doi.org/10.1007/s11295-017-1126-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1126-z