
Abstract
Soluble sugars and organic acids have a strong impact on the overall organoleptic quality of fruits. In this study, we report the identification of quantitative trait loci (QTLs) for individual sugars and organic acids in apple. A high density linkage map of apple was constructed using the 1536 EST-derived SNP GoldenGate genotyping platform. The linkage map consists of 601 molecular markers, including 540 single nucleotide polymorphisms (SNPs) and 61 simple sequence repeats (SSRs), spanning 1368.4 cM with an average of 2.28 cM per marker. The contents of soluble sugars, including sucrose, glucose, fructose, sorbitol, and organic acids, including malic acid and citric acid, were used as the phenotypic data in QTL analysis. Two QTLs for malic acid content were detected on linkage groups (LGs) 8 and 16, while no QTL was found for citric acid content. Four QTLs for the glucose, sucrose, fructose, and sorbitol content were found to be clustered in one region on LG 3. Moreover, an additional QTL for glucose content was detected on the LG 4. Our study not only expands our understanding of the genetic basis for fruit organoleptic quality but it also provides molecular markers that will aid in marker-assisted selection for fruit quality in apple breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apple (Malus×domestica Borkh.) is one of the most important temperate fruit crops in the world with the cultivation history dating as early as 1000 BC (Zohary and Hopf 2000). Apple breeding is difficult and time-consuming due to its self-incompatibility, long juvenile period, and high heterozygosity. To save time and reduce cost in apple breeding program, marker-assisted selection (MAS) is a potential process in identifying young seedlings with favorable alleles or desirable traits (Longhi et al. 2014). Thus, developing molecular markers close to candidate gene(s) or quantitative trait loci (QTLs) which control economically desired traits is an important goal in the apple breeding program. In the past couple of decades, a variety of QTLs or major genes have been identified which control important horticultural traits, such as disease resistance (Xu and Korban 2002; Belfanti et al. 2004; Calenge and Durel 2006; Le Roux et al. 2010; Gardiner et al. 2012), tree growth and architecture (Kenis and Keulemans 2007; Segura et al. 2008; Bai et al. 2012a; Kumar et al. 2013), fruit quality (Espley et al. 2009; Mellidou et al. 2012; Longhi et al. 2012; Khan et al. 2012a; Zhang et al. 2012; Longhi et al. 2013; Di Guardo et al. 2013; Wu et al. 2014), bud dormancy, and bud break (Brunel et al. 2002; Graham et al. 2009; van Dyk et al. 2010).
Linkage map (also known as genetic map) provides a framework of marker order and spacing, and it serves as a starting point to identify QTLs of interest. In apple, several linkage maps have been constructed with molecular markers such as amplified fragment length polymorphisms (AFLPs), random amplified polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP), and simple sequence repeats (SSRs) using a two-way pseudo-testcross mapping strategy (Maliepaard et al. 1998; Liebhard et al. 2003a; Han et al. 2011; Zhang et al. 2012). However, the marker density of these linkage maps is usually low and insufficient for fine mapping of functional genetic variation. With the development of high-throughput DNA sequencing technologies, single nucleotide polymorphisms (SNPs), the most common type of DNA sequence variation in the genome, were explored by whole-genome resequencing to develop SNP array in apple (Chagné et al. 2012; Bianco et al. 2014; Gardner et al. 2014). Recently, we explored SNPs by comparison of expressed-sequence tag (EST) data that were generated from 14 apple genotypes and a total of 1536 SNPs were selected to develop a GoldenGate™ array (Khan et al. 2012c). A couple of high-density SNP-based linkage maps were successfully constructed using SNP arrays in apple (Antanaviciute et al. 2012; Khan et al. 2012b; Falginella et al. 2015). The high-density linkage map will facilitate the identification of candidate genes of interest embedded in the assembled sequence data of the apple genome (Velasco et al. 2010).
Improvement of fruit quality is an important goal in all apple breeding programs. Soluble sugars and organic acids are important components of fruit taste, and they exert a strong impact on the overall organoleptic quality of fruits (Borsani et al. 2009). Soluble sugars in fruits are mainly composed of sucrose, fructose, and glucose, while malic, citric, and tartaric acids are the primary organic acids. Different sugar or organic acid components differ in sweetness or acidity (Doty 1976); thus, organoleptic fruit quality (or fruit taste) depends on the content and type of soluble sugars and organic acids. Several QTLs for fruit acidity in apple have been reported (Maliepaard et al. 1998; Liebhard et al. 2003b; Kenis et al. 2008; Zhang et al. 2012; Xu et al. 2012), and a major QTL (also called as the Ma locus) located on the top of linkage group (LG) 16 was successfully isolated (Bai et al. 2012b; Khan et al. 2013; Ma et al. 2015a). Likewise, more than 10 QTLs for fruit sweetness have been detected, but with no major effect (Liebhard et al. 2003b; Guan et al. 2015). However, in previous QTL mapping studies, soluble solids and pH values or titratable acidity were commonly used as phenotypic data. Our understanding and knowledge of the genetic basis of the components and contents of soluble sugars and organic acids in apple fruits are still limited.
In this study, a dense genetic map was constructed using the 1536 EST-derived SNP GoldenGate genotyping platform and an F1 segregating population. The composition and content of soluble sugars and organic acids in mature fruits of the segregating population were quantified using high-performance liquid chromatography (HPLC). The aim of this study is to identify QTLs for the component and content of soluble sugars and organic acids in apple fruit. The results will not only expand our understanding of the mechanisms underlying fruit taste quality but will also aid in map-based isolation of genes responsible for soluble sugars or organic acid contents in apple fruit.
Materials and methods
Plant material
An apple segregating F1 population derived from a cross between ‘Jiguan (JG)’ and ‘Wangshanhong (WSH),’ maintained at Liaoning Institute of Pomology (Xiongyue, Liaoning Province, China), was used for linkage map construction and QTL analysis. The population consists of 191 individuals. Young leaves of each individual along with parents were collected for genome DNA extraction at spring season. DNA extraction was conducted using the Plant Genomic DNA Kit (TianGen, Beijing, China) according to the manufacturer’s instructions, and DNA quality and concentration were detected using NanoDrop™ Lite Spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Fruits were collected at maturity and the maturity was assessed by checking the color of the peel and a confirmation of the seed color changing to brown. Three biological replicates from each individual were prepared, and each consisted of 10 fruits. The fruits were mechanically peeled and pulps were cut into small pieces. The fruit samples were immediately frozen in liquid nitrogen, and then stored at −40 °C for soluble sugars and organic acid content measurement by high-performance liquid chromatography.
Measurement of sugar and organic acid content
The fruit samples were ground into a fine powder in liquid nitrogen and 1 g of powder was dissolved in 6 mL deionized water that was generated by a Milli-Q Element water purification system (Millipore, Bed ford, MA, USA). The mixture was centrifuged at 5000×g for 15 min at 4 °C and the supernatants were filtered through a SPE-C18 cartridge (Supelclean ENVI C18 SPE) and a 0.22-μm millipore membrane. The filtered fluid was used to measure soluble sugar and organic acid contents using a Dionex P680 HPLC system (Dionex Corporation, CA, USA). The refractive index detector was used to measure fructose, glucose, sucrose, and sorbitol content. Degassed, distilled, deionized water at a flow rate of 0.6 mL/min was used as the mobile phase. The total injection volume was 20 μL. Organic acids, including malic acid and citric acid, were detected using an ultraviolet detector. Samples were eluted with 0.02 mol/L KH2PO4 solution (pH 2.4). The flow rate was 0.8 mL/min. Eluted compounds were detected by UV absorbance at 210 nm. Soluble sugars and organic acid concentrations were expressed in milligram per gram fresh weight (FW).
Analysis of SNP and SSR genotyping
The SNP genotyping was conducted using the Illumina 1536 GoldenGate™ assay on the BeadStation system (Illumina Inc., San Diego, CA) at the W. M. Keck Center for Functional Genomics (University of Illinois at Urbana-Champaign), according to our previously reported protocol (Khan et al. 2012c). Briefly, a total of 250 ng genomic DNA each individual was used for SNP genotyping. The fluorescence images of an array matrix carrying Cy3- and Cy5-labeled beads were generated with the two-channel scanner. The normalization procedure was carried out using the genotyping function in the BeadStudio package (Illumina, San Diego, CA), and genotype calls and clustering were generated using the GenCall (GC) function of the BeadStudio package. The GC score ≥0.35 for each SNP was set as the threshold to define a successful genotyping call. SNPs with average GC scores lower than 0.35 were deemed as failures and were thus removed from the analysis.
For SSR genotyping, PCR reaction was performed in a GeneAmp PCR System 9700 (ABI, USA) using the following cycling protocol: 5 min at 95 °C, followed by 35 cycles consisting of 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and with a final extension of 72 °C for 10 min. Three microliters of amplicons was mixed with an equal volume of formamide loading buffer (98 % formamide, 10 mM EDTA, pH 8.0, 0.025 % bromophenol blue, and xylene cyanol). The mixture was denatured at 94 °C for 3 min, and then immediately chilled on ice. An aliquot of 2 μL mixture was analyzed on 8 % denaturing polyacrylamide gel for 1–1.5 h at 1200 V. Bands were visualized after silver staining and their sizes were estimated according to a standard 25 bp DNA ladder standard (Promega, Madison, WI).
Linkage group construction and QTL analysis
The F1 population mentioned above were used to construct genetic linkage maps using JoinMap version 4.0 software (Van Ooijen 2004), as previously described by Han et al. (2011) and Zhang et al. (2012). The Kosambi mapping function was used to calculate the genetic distance between molecular markers as described by Liebhard et al. (2003a), map distances were denoted in centimorgan. A logarithm of odds (LOD) of 4.0 was used to assign markers to different linkage groups. Linkage maps were drawn using MapChart for Windows (Voorrips 2002). The genetic maps for each parent were initially constructed, and a consensus map was subsequently built using the CP population model. Order of markers in genetic maps for each parent was used as preferred orders for the construction of the consensus map.
QTL analysis was performed with the MapQTL 5.0 software (Van Ooijen 2006). The Kruskal-Wallis (KW) test was used to detect the candidate QTLs for the qualitative data, and the Interval Mapping (IM) was used to identify QTLs in the quantitative data. The nearby loci with the highest LOD scores were selected as co-factors for Multiple QTL model (MQM) computation. The significant threshold was computed using a 1000 permutation test, at the 95 % genome-wide LOD thresholds.
Results
Variation in soluble sugar and organic acid contents among the segregating population
Soluble sugars (sucrose, glucose, fructose, and sorbitol) and organic acids (malic acid and citric acid) in mature fruits of the JG×WSH segregating population were quantified using HPLC and the distribution was shown in Fig. 1. The content of three sugar components, sucrose, fructose, and glucose, showed a normal distribution, whereas the distribution of sorbitol content was slightly skewed towards low sorbitol contents. The average concentrations of sucrose, fructose, glucose, and sorbitol were 3.62, 59.54, 12.75, and 3.41 mg/g FW, respectively (Table 1). This indicates that fructose and glucose are the major sugar components in apple mature fruit.

Distribution of soluble sugar and organic acid contents in mature fruits of the JG×WSH mapping population. The arrows indicate the phenotypic values of parents
The malic acid content showed a normal distribution, while the distribution of citric acid content was skewed towards low citric acid contents. The average content of malic acid in mature apple fruit was 4.28 mg/g FW, ranging from 1.31 to 11.58 mg/g FW (Table 1). In contrast, the average content of citric acid in mature apple fruit was as low as 0.12 mg/g FW, ranging from undetectable level to 1.11 mg/g FW. These results demonstrate that malic acid is the predominant organic acid in apple mature fruit. The ratio of malic acid content to the total organic acid content ranged from 81.62 to 100.00 %, with an average of 97.3 % (Fig. S1).
SNP and SSR genotyping of the segregating population
Genotyping of the segregating F1 population was initially conducted using the Illumina GoldenGate™ assay. Of the 1536 SNPs, 806 (52.5 %) had valid GoldenGate assay scores. Later, 189 SNPs with no segregation information in the F1 population (segregation type of aa×aa, aa×bb, bb×aa, and bb×bb) were discarded, resulting in a total of 617 valid SNPs with segregation type of ef×eg (1.8 %), hk×hk (23.2 %), lm×ll (38.9 %), and nn×np (36.1 %). These valid SNPs were classified into four categories according to their polymorphisms, designated type I, type II, type III, and type IV. Type I represented A/C or T/G transversions, and type II indicated A/G or T/C transversions. Types III and IV represented A/T and C/G transitions, respectively. Type II was the most abundant and accounted for 60.9 % of all the valid SNPs, while types I, III, and IV accounted for 13.0, 18.0, and 8.1 %, respectively.
A total of 162 SSR markers were initially used to screen the parents ‘JG’ and ‘WSH.’ Of the 162 SSR markers, 102 were selected from a public domain of apple molecular markers (http://www.hidras.unimi.it/), and 60 with a ‘WBGCAS’ prefix were recently developed in our laboratory (Zhang et al. 2012). As a result, 61 SSRs were identified to be polymorphic between ‘JG’ and ‘WSH,’ and were then used to genotype the F1 population. Microsatellite genotyping revealed five segregation types, i.e., lm×ll, nn×np, hk×hk, ab×cd, and ef×eg.
Construction of linkage groups of apple
The 620 valid SNPs and 61 SSRs mentioned above were initially used to construct linkage groups. However, 80 SNPs were subsequently excluded because either they failed to link with any linkage groups or their distorted segregation conflicted with segregation patterns of neighboring markers. In addition, 36 seedlings were excluded from late linkage analysis as they exhibited several double recombination events. As a result, a consensus linkage map, which contained 601 loci along 17 linkage groups, was finally constructed (Fig. S2). The number of molecular markers on each linkage group ranged from 23 (LG 10) to 47 (LG 3 and LG 11), with an average of 35.4 (Table 2). Linkage group 10 had the fewest number of markers, with 23 markers spanning a genetic distance of 82.0 cM. Whereas, linkage groups 3 and 11 had the largest number of markers, with 47 markers spanning a genetic distance of 80.2 and 72.5 cM, respectively. Linkage group 8 contained 30 molecular markers and had the lowest average density of 3.72 cM per marker, while linkage group 12 consisted of 44 molecular markers and had the highest average density of 1.36 cM per marker. This suggests that LGs 8 and 12 have the lowest and highest rates of heterozygosity, respectively. With the exception of linkage groups 8 and 12, the marker density of the linkage groups ranged from 1.54 cM per marker (LG 11) to 3.56 cM per marker (LG 10).
The genetic length of each linkage group ranged from 59.2 cM (LG 12) to 120.0 cM (LG 8). The consensus linkage map spanned 1368.4 cM, with an average density of 2.28 cM per marker. Of the 601 markers along the consensus maps, 256 and 227 were mapped onto the linkage maps of ‘Wangshanhong’ and ‘Jiguan,’ respectively. The total lengths of linkage maps of ‘Wangshanhong’ and ‘Jiguan’ were 1114.8 and 1225.5 cM, respectively, and the average densities were 4.35 and 5.40 cM per marker, respectively.
Indentifying QTLs for soluble sugar and organic acids contents in mature apple fruit
QTL analysis was conducted for the content of soluble sugars, including sucrose, fructose, glucose, and sorbital, and organic acids, including malic acid and citric acid, using the Kruskal–Wallis test with a significance level of 0.01 and interval mapping and multiple QTL model with a threshold LOD score of 2.8. As a result, a total of seven QTLs were identified (Table 3). Among those QTLs, one QTL was detected for sucrose content, two QTLs for the glucose content, one QTL for fructose content, one QTL for sorbitol content, and two QTLs for malic acid content. Interestingly, one QTL cluster responsible for the content of sucrose, glucose, fructose, and sorbitol was detected in the middle region of linkage group 3 (Fig. 2). Besides this QTL cluster, another QTL for glucose content was identified on LG4. Two QTLs for malic acid content were located on linkage groups 8 and 16, respectively.

QTLs for the content of soluble sugars and organic acids detected in the JG×WSH mapping population. LG, linkage group; F, female; M, male. The genetic distance is shown in centimorgan on the left side. The confidence interval for QTL location is indicated by black vertical line
The major QTL for sucrose content on LG 3 accounted for 11.7 % of the phenotypic variation, with a peak LOD score of 3.41. The two major QTLs for glucose content on LGs 3 and 4 accounted for 17.1 and 16.7 % of the phenotypic variation, with peak LOD scores of 4.47 and 4.49, respectively. The major QTL for fructose content on LG 3 accounted for 20.6 % of the phenotypic variation, with a peak LOD score of 5.75. The major QTL for sorbitol content on LG 3 had a peak LOD score of 7.72 and accounted for 28.0 % of the phenotypic variation.
The two major QTLs for malic acid content on LGs 8 and 16 accounted for 14.4 and 12.4 % of the phenotypic variation, with peak LOD scores of 4.0 and 3.42, respectively. The major QTL on linkage group 8 was flanked by two markers MdSNPui10630 and Hi01f09, which is consistent with our previous study (Zhang et al. 2012). The major QTL on top of LG 16 was flanked by two markers WBGCAS149-1 and MdSNPui10630, which is consistent with previous reports (Maliepaard et al. 1998; Liebhard et al. 2003b; Kenis et al. 2008).
All the QTLs for the content of soluble sugars were detected on the linkage groups of ‘Wangshanhong.’ In contrast, all the QTLs for malic acid were detected on the linkage groups of ‘Jiguan.’ In addition, we also checked statistical difference in average phenotypic value between individuals carrying different genotypes based on SNPs closest to the QTL peaks (Fig. 3). MdSNPui08437 was closest to the QTL cluster on chromosome 3 and its genotyping result revealed two groups in the segregating population. The individuals carrying the homozygous nn genotype exhibited significantly lower concentrations of sugar components in fruit compared with the individuals carrying the heterozygous np genotype. MdSNPui05013 was closest to the QTL peak on chromosome 4 and individuals carrying on different genotypes at this SNP locus showed significant difference in glucose content in fruit. Likewise, MdSNPui10630 and MdSNPui08981 were closest to the QTL peaks on chromosomes 8 and 16, respectively, and they both generated two genotypes, ll and lm. The individuals carrying the homozygous ll genotype exhibited significant difference in malic acid content in fruit compared with the individuals carrying the heterozygous lm genotype.

Phenotypic distribution of soluble sugar and malic acid contents measured in the ‘JG’בWSH’ segregating population based on SNPs closest to the QTL peaks. Capital letters, A, B, C, and D, indicate QTLs for fruit organoleptic quality on chromosomes 3, 4, 8, and 16, respectively. Asterisks “*” and “**” indicate significance levels at P < 0.05 and P < 0.01, respectively
Discussion
SSRs and SNPs have been widely used to construct high-density linkage map in apple (Han et al. 2011; Zhang et al. 2012; Khan et al. 2012b). In this study, a linkage map was developed using 601 molecular markers, including 61 SSRs and 540 SNPs. Mapping results of these SSRs and SNPs were compared with both their positions along the draft genome of apple and previously reported linkage maps. Of the 61 SSRs, 60 were mapped on the same linkage groups as previously reported (Liebhard et al. 2002) and their genetic map positions were consistent with sequence-based physical-map positions. One gSSR, CH02b10, was previously mapped onto LG2 (Liebhard et al. 2002). However, our study revealed for the first time that it was genetically mapped onto two loci on LG2. For the 540 SNPs, 483 were consistent between their genetic map positions and sequence-based physical-map positions, while 57 were inconsistent (Table S2). Of these 57 SNPs, 30 and 6 were mapped onto the same or different linkage groups, respectively, as previously reported (Khan et al. 2012b, 2014). Twenty-one were mapped for the first time onto linkage groups. These results suggest that our linkage map is likely to be accurate and the inconsistency as mentioned above may be attributed to structural variations in the apple genome (Khan et al. 2012b; Han et al. 2011).
Apple has a long juvenile phase of 5–12 years. Selecting for organoleptic quality in apple by marker-assisted selection at the young seedling stage has the potential to save time and space and reduce the cost for breeding programs. Organic acids and soluble sugars play a critical role in determining fruit organoleptic quality. Several QTLs for fruit sweetness and acidity have been identified in apple (Maliepaard et al. 1998; Liebhard et al. 2003b; Kenis et al. 2008; Zhang et al. 2012; Xu et al. 2012; Guan et al. 2015). For acidity in apple, the QTL studies were mainly conducted for titratable acidity (TA) and/or pH, and two QTLs were consistently detected across different studies. One is a major QTL on top of LG 16 (also known as the Ma locus) (Maliepaard et al. 1998; Liebhard et al. 2003b; Liebhard et al. 2003b), and another is located on LG 8 (Liebhard et al. 2003b; Kenis et al. 2008; Zhang et al. 2012). In this study, QTL mapping was conducted for individual acids and two QTLs for malic acid were detected on LGs 8 and 16. Our result is consistent with the result of QTL mapping for TA and/or pH (Maliepaard et al. 1998; Liebhard et al. 2003b; Kenis et al. 2008; Xu et al. 2012). This consistency is most likely due to the fact that malic acid is the principal acid and accounts for approximately 90 % of total organic acids in apple fruit (Wu et al. 2007; Ma et al. 2015b).
Our study reveals that apple fruit accumulates a small amount of citric acid and its proportion can reach as high as 18.4 % at harvest. However, no QTLs for citric acid were detected. This is likely due to the following: (1) the two parents of the mapping population both accumulate very low levels of citrate in fruits; (2) citrate accumulation is influenced by the interaction between genotype and environment, resulting in the mapping population segregating for citrate content in fruits. In addition, uptake of citrate into vacuolar occurs possibly through the malate channel (Rentsch and Martinoia 1991), but the malate channel transports citrate much more slowly than malate (Hafke et al. 2003). Thus, citrate accumulation in fruit cells is believed to be primarily controlled by its metabolism as high concentrations of cytosolic citrate can increase the transport capacity of citrate into the vacuole (Etienne et al. 2013). Citrate synthase (CS) directly controls citrate synthesis, while aconitase (ACO) and enzymes such as NAD-dependant isocitrate dehydrogenase (NAD-IDH) are involved in citrate degradation (Sadka et al. 2001; Wen et al. 2001; Iannetta et al. 2004). Therefore, it is worthy of further study to ascertain if genes encoding enzymes such as CS, ACO, and NAD-IDH are responsible for the extremely low level of citric acid in apple fruit.
Several studies show that genes encoding malate dehydrogenase and malic enzyme may play an important role in mediating the accumulation of malate in apple fruit (Chen et al. 2009; Sweetman et al. 2009; Yao et al. 2011). However, increasing evidence indicates that malate accumulation in fruit cells is mainly controlled at the level of vacuolar storage (Verweij et al. 2008; Etienne et al. 2013). This hypothesis is proved by the recent finding that the major QTL Ma was shown to encode an aluminium-activated malate transporter that functions in transport of malic acid into vacuole (Bai et al. 2012b; Khan et al. 2013; Ma et al. 2015a).
Previous QTL studies for apple sweetness mainly focus on soluble solid content (SSC) as sugars are the major soluble solid in fruit juice. To date, QTL mapping for SSC was conducted in five different F1 segregating populations and QTLs were detected on LGs 2, 3, 6, 8, 9, 10, 12, 13, 14, 15, and 16 (Liebhard et al., 2003b; Kenis et al. 2008; Kunihisa et al. 2014; Potts et al. 2014; Guan et al. 2015). However, no QTL was consistently detected across these mapping populations. The inconsistency is most likely due to the following: (1) SSC is a trait with complex composition, including not only the individual sugars but also organic acids, amino acids, phenolic compounds, and soluble pectins (Young et al. 1993; Kader 2008); (2) SSC is a quantitative trait highly influenced by environment (Alspach and Oraguzie 2002; Inglese et al. 2010); and (3) the different mapping populations were used.
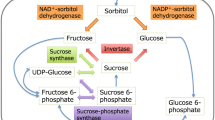
Since the composition of soluble solids is complex, several studies have reported QTL mapping for individual sugars in apple. Guan et al. (2015) detected a QTL cluster on LG 1 for the contents of fructose, glucose, sucrose, and sorbitol, while Kunihisa et al. (2014) reported a QTL cluster on LG 16 for fructose and sorbitol contents. In this study, four QTLs for the glucose, sucrose, fructose, and sorbitol content were found to be clustered in one region on LG 3 from 48.60 to 50.60 cM. The clustering QTLs for individual sugars were also discovered in peach (Dirlewanger et al. 1999; Etienne et al. 2002). These interesting clusters are likely caused by the tightly linked genes for individual sugars or the pleiotropic effect of a single gene encoding a key enzyme involved in the glycometabolism (Guan et al. 2015). For example, invertase could be such a key enzyme as it converts sucrose into glucose and fructose. Moreover, soluble sugars are mainly stored in the vacuoles of fruit cells. More recently, two members of the SWEET family (Sugar Will Eventually Be Exported Transporter), AtSWEET 16 and AtSWEET 17, reside in the tonoplast and transport glucose, fructose, and sucrose in Arabidopsis (Klemens et al. 2013; Chardon et al. 2013). Thus, genes encoding sugar transporter could be likely candidate QTLs for individual sugar accumulation in fruits such as apple. In addition, correlation between sugar components was frequently reported in apple fruit (Paterson et al. 1991; Kunihisa et al. 2014; Guan et al. 2015; Ma et al. 2015b), and the strong correlation between the sugar components is also an important reason for forming QTL cluster (Paterson et al. 1991). In this study, significant positive correlation was observed between soluble sugar components (Table S1), which may be partially attributed to gene(s) with pleiotropic effect on sugar metabolism or the cluster of genes/QTLs for individual sugars.
References
Alspach PA, Oraguzie NC (2002) Estimation of genetic parameters of apple (Malus×domestica) fruit quality from open-pollinated families. N Z J Crop Hortic Sci 30:219–228
Antanaviciute L, Fernández-Fernández F, Jansen J, Banchi E, Evans KM, Viola R, Velasco R, Dunwell JM, Troggio M, Sargent DJ (2012) Development of a dense SNP-based linkage map of an apple rootstock progeny using the Malus Infinium whole genome genotyping array. BMC Genomics 13:203
Bai T, Zhu Y, Fernández-Fernández F, Keulemans J, Brown S, Xu K (2012a) Fine genetic mapping of the Co locus controlling columnar growth habit in apple. Mol Genet Genomics 287:437–450
Bai Y, Dougherty L, Li M, Fazio G, Cheng L, Xu K (2012b) A natural mutation-led truncation in one of the two aluminum-activated malate transporter-like genes at the Ma locus is associated with low fruit acidity in apple. Mol Genet Genomics 287:663–678
Belfanti E, Silfverberg-Dilworth E, Tartarini S, Patocchi A, Barbieri M, Zhu J, Vinatzer BA, Gianfranceschi L, Gessler C, Sansavini S (2004) The HcrVf2 gene from a wild apple confers scab resistance to a transgenic cultivated variety. Proc Natl Acad Sci 101:886–890
Bianco L, Cestaro A, Sargent DJ, Banchi E, Derdak S, Di Guardo M, Salvi S, Jansen J, Viola R, Gut I, Laurens F, Chagné D, Velasco R, van de Weg E, Troggio M (2014) Development and validation of a 20K single nucleotide polymorphism (SNP) whole genome genotyping array for apple (Malus×domestica Borkh). PLoS One 9:e110377
Borsani J, Budde CO, Porrini L, Lauxmann MA, Lombardo VA, Murray R, Andreo CS, Drincovich MF, Lara MV (2009) Carbon metabolism of peach fruit after harvest: changes in enzymes involved in organic acid and sugar level modifications. J Exp Bot 60:1823–1837
Brunel N, Leduc N, Poupard P, Simoneau P, Mauget JC, Viémont JD (2002) KNAP2, a class I KN1-like gene is a negative marker of bud growth potential in apple trees (Malus domestica [L.] Borkh.). J Exp Bot 53:2143–2149
Calenge F, Durel CE (2006) Both stable and unstable QTLs for resistance to powdery mildew are detected in apple after four years of field assessments. Mol Breed 17:329–339
Chagné D, Crowhurst RN, Troggio M, Davey MW, Gilmore B, Lawley C, Vanderzande S, Hellens RP, Kumar S, Cestaro A, Velasco R, Main D, Rees JD, Iezzoni A, Mockler T, Wilhelm L, Van de Weg E, Gardiner SE, Bassi N, Peace C (2012) Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS One 7:e31745
Chardon F, Bedu M, Calenge F, Klemens PAW, Spinner L, Clement G, Chietera G, Léran S, Ferrand M, Lacombe B, Loudet O, Dinant S, Bellini C, Neuhaus HE, Daniel-Vedele F, Krapp A (2013) Leaf fructose content is controlled by the vacuolar transporter SWEET17 in Arabidopsis. Curr Biol 23:697–702
Chen FX, Liu XH, Chen LS (2009) Developmental changes in pulp organic acid concentration and activities of acid-metabolising enzymes during the fruit development of two loquat (Eriobotrya japonica Lindl.) cultivars differing in fruit acidity. Food Chem 114:657–664
Dirlewanger E, Moing A, Rothan C, Svanella L, Pronier V, Guye A, Plomion C, Monet R (1999) Mapping QTLs controlling fruit quality in peach (Prunus persica (L.) Batsch). Theor Appl Genet 98:18–31
Di Guardo M, Tadiello A, Farneti B, Lorenz G, Masuero D, Vrhovsek U, Costa G, Velasco R, Costa F (2013) A multidisciplinary approach providing new insight into fruit flesh browning physiology in apple (Malus x domestica Borkh.). PLos one 8:e78004
Doty TE (1976) Fructose sweetness: a new dimension. Cereal Foods World 21:62–63
Etienne C, Rothan C, Moing A, Plomion C, Bodénès C, Svanella-Dumas L, Cosson P, Pronier V, Monet R, Dirlewanger E (2002) Candidate genes and QTLs for sugar and organic acid content in peach [Prunus persica (L.) Batsch]. Theor Appl Genet 105:145–159
Etienne A, Génard M, Lobit P, Mbeguié-A-Mbéguié D, Bugaud C (2013) What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J Exp Bot 64:1451–1469
Espley RV, Brendolise C, Chagné D, Kutty-Amma S, Green S, Volz R, Putterill J, Schouten HJ, Gardiner SE, Hellens RP, Allan AC (2009) Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 21:168–183
Falginella L, Cipriani G, Monte C, Gregori R, Testolin R, Velasco R, Troggio M, Tartarini S (2015) A major QTL controlling apple skin russeting maps on the linkage group 12 of ‘Renetta Grigia di Torriana’. BMC Plant Biol 15:150
Gardiner SE, Norelli JL, de Silva N, Fazio G, Peil A, Malnoy M, Horner M, Bowatte D, Carlisle C, Wiedow C, Wan Y, Bassett CL, Baldo AM, Celton JM, Richter K, Aldwinckle HS, Bus VG (2012) Putative resistance gene markers associated with quantitative trait loci for fire blight resistance in Malus ‘Robusta 5’ accessions. BMC Genet 13:25
Gardner KM, Brown P, Cooke TF, Cann S, Costa F, Bustamante C, Velasco R, Troggio M, Myles S (2014) Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3 4:1681–1687
Graham J, Hackett CA, Smith K, Woodhead M, Hein I, McCallum S (2009) Mapping QTLs for developmental traits in raspberry from bud break to ripe fruit. Theor Appl Genet 118:1143–1155
Guan Y, Peace C, Rudell D, Verma S, Evan K (2015) QTLs detected for individual sugars and soluble solids content in apple. Mol Breed 35:135
Hafke JB, Hafke Y, Smith JAC, Lüttge U, Thiel G (2003) Vacuolar malate uptake is mediated by an anion-selective inward rectifier. Plant J 35:116–128
Han YP, Zheng D, Vimolmangkang S, Khan MA, Beever JE, Korban SS (2011) Integration of physical and genetic maps in apple confirms whole genome and segmental duplications in the apple genome. J Exp Bot 62:5117–5130
Iannetta PPM, Escobar NM, Ross HA, Souleyre EJF, Hancock RD, Witte C-P, Davies HV (2004) Identification, cloning and expression analysis of strawberry (Fragaria×ananassa) mitochondrial citrate synthase and mitochondrial malate dehydrogenase. Physiol Plant 121:15–26
Inglese P, Costanza P, Gugliuzza G, Inglese G, Liguori G (2010) Influence of within-tree and environmental factors on fruit quality of cactus pear (Opuntia ficus-indica) in Italy. Fruits 65:179–189
Kader AA (2008) Flavor quality of fruits and vegetables. J Sci Food Agric 88:1863–1868
Kenis K, Keulemans J (2007) Study of tree architecture of apple (Malus × domestica Borkh.) by QTL analysis of growth traits. Mol Breeding 19:193–208
Kenis K, Keulemans J, Davey MW (2008) Identification and stability of QTLs for fruit quality traits in apple. Tree Genet Genomes 4:647–661
Khan SA, Chibon PY, de Vos RC, Schipper BA, Walraven E, Beekwilder J, van Dijk T, Finkers R, Visser RG, van de Weg EW, Bovy A, Cestaro A, Velasco R, Jacobsen E, Schouten HJ (2012a) Genetic analysis of metabolites in apple fruits indicates an mQTL hotspot for phenolic compounds on linkage group 16. J Exp Bot 63:2895–2908
Khan MA, Han Y, Zhao YF, Troggio M, Korban SS (2012b) A multi-population consensus genetic map reveals inconsistent marker order among maps likely attributed to structural variations in the apple genome. PLoS One 7:e47864
Khan MA, Han Y, Zhao YF, Korban SS (2012c) A high-throughput apple SNP genotyping platform using the GoldenGate™ assay. Gene 494:196–201
Khan MA, Olsen KM, Sovero V, Kushad MM, Korban SS (2014) Fruit quality traits have played critical roles in domestication of the apple. Plant Genome 7:1–18
Khan SA, Beekwilder J, Schaart JG, Mumm R, Soriano JM, Jacobsen E, Schouten HJ (2013) Differences in acidity of apples are probably mainly caused by a malic acid transporter gene on LG16. Tree Genet Genomes 9:475–487
Klemens PAW, Patzke K, Deitmer J, Spinner L, Hir RL, Bellini C, Bedu M, Chardon F, Krapp A, Neuhaus HE (2013) Overexpression of the vacuolar sugar carrier AtSWEET16 modifies germination, growth, and stress tolerance in Arabidopsis. Plant Physiol 163:1338–1352
Kumar S, Garrick DJ, Bink MC, Whitworth C, Chagné D, Volz RK (2013) Novel genomic approaches unravel genetic architecture of complex traits in apple. BMC Genomics 14:393
Kunihisa M, Moriya S, Abe K, Okada K, Haji T, Hayashi T, Kim H, Nishitani C, Terakami S, Yamamoto T (2014) Identification of QTLs for fruit quality traits in Japanese apples: QTLs for early ripening are tightly related to preharvest fruit drop. Breed Sci 64:240–251
Le Roux PM, Khan MA, Broggini GA, Duffy B, Gessler C, Patocchi A (2010) Mapping of quantitative trait loci for fire blight resistance in the apple cultivars ‘Florina’ and ‘Nova Easygro’. Genome 53:710–722
Liebhard R, Gianfranceschi L, Koller B, Ryder CD, Tarchini R, Van De Weg E, Gessler C (2002) Development and characterisation of 140 new microsatellites in apple (Malus x domestica Borkh.). Mol Breed 10:217–241
Liebhard R, Koller B, Gianfranceschi L, Gessler C (2003a) Creating a saturated reference map for the apple (Malus x domestica Borkh.) genome. Theor Appl Genet 106:1497–1508
Liebhard R, Kellerhals M, Pfammatter W, Jertmini M, Gessler C (2003b) Mapping quantitative physiological traits in apple (Malus × domestica Borkh.). Plant Mol Biol 52:511–526
Longhi S, Giongo L, Buti M, Surbanovski N, Viola R, Velasco R, Ward JA, Sargent DJ (2014) Molecular genetics and genomics of the Rosoideae: state of the art and future perspectives. Hortic Res 1:1
Longhi S, Moretto M, Viola R, Velasco R, Costa F (2012) Comprehensive QTL mapping survey dissects the complex fruit texture physiology in apple (Malus x domestica Borkh.). J Exp Bot 63:1107–1121
Longhi S, Hamblin MT, Trainotti L, Peace CP, Velasco R, Fabrizio C (2013) A candidate gene based approach validates Md-PG1 as the main responsible for a QTL impacting fruit texture in apple (Malus × domestica Borkh). BMC Plant Biol 13:37
Ma BQ, Liao L, Zheng HY, Chen J, Wu BH, Ogutu C, Li SH, Korban SS, Han YP (2015a) Genes encoding aluminum-activated malate transporter II and their association with fruit acidity in apple. Plant Genome 8: doi: 10.3835
Ma BQ, Chen J, Zheng HY, Fang T, Collins O, Li SH, Han YP, Wu BH (2015b) Comparative assessment of sugar and malic acid composition in cultivated and wild apples. Food Chem 172:86–91
Maliepaard C, Alston FH, van Arkel G, Brown LM, Chevreau E, Dunemann KM, Gaediner S, Guilford P, van Heusden AW, Janse J, Laurens F, Lynn JR, Manganaris AG, den Nijs APM, Periam N, Rikkerink E, Roche P, Ryder C, Sansavini S, Schmidt H, Tartarini S, Verhaegh JJ, Vrielink-van Ginkel M, King GJ (1998) Aligning male and female linkage maps of apple (Malus pumila Mill.) using multi-allelic markers. Theor Appl Genet 97:60–73
Mellidou I, Chagné D, Laing WA, Keulemans J, Davey MW (2012) Allelic variation in paralogs of GDP-L-galactose phosphorylase is a major determinant of vitamin C concentrations in apple fruit. Plant Physiol 160:1613–1629
Paterson AH, Damon S, Hewitt JD, Zamir D, Rabinowith HD, Lincoln SE, Lander ES, Tanksley SD (1991) Mendelian factors underlying quantitative traits in tomato: comparison across species, generations, and environments. Genetics 127:181–197
Potts SM, Khan MA, Han Y, Kushad MM, Korban SS (2014) Identification of quantitative trait loci (QTLs) for fruit quality traits in apple. Plant Mol Biol Report 32:109–116
Rentsch D, Martinoia E (1991) Citrate transport into barley mesophyll vacuoles—comparison with malate uptake activity. Planta 184:532–537
Sadka A, Dahan E, Or E, Roose ML, Marsh KB, Cohen L (2001) Comparative analysis of mitochondrial citrate synthase gene structure, transcript level and enzymatic activity in acidless and acid-containing Citrus varieties. Funct Plant Biol 28:383–390
Segura V, Cilas C, Costes E (2008) Dissecting apple tree architecture into genetic, ontogenetic and environmental effects: mixed linear modelling of repeated spatial and temporal measures. New Phytol 178:302–314
Sweetman C, Deluc LG, Cramer GR, Ford CM, Soole KL (2009) Regulation of malate metabolism in grape berry and other developing fruits. Phytochemistry 70:1329–1344
van Dyk MM, Soeker MK, Labuschagne IF, Rees DJG (2010) Identification of a major QTL for time of initial vegetative budbreak in apple (Malus × domestica Borkh.). Tree Genet Genomes 6:489–502
Van Ooijen JW (2004) MapQTL® 5.0, software for the mapping of quantitative trait loci in experimental populations. Kyazma BV, Wageningen
Van Ooijen JW (2006) JoinMapW 4, software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D, Salvi S, Pindo M, Baldi P, Castelletti S, Cavaiuolo M, Coppola G, Costa F, Cova V, Dal Ri A, Goremykin V, Komjanc M, Longhi S, Magnago P, Malacarne G, Malnoy M, Micheletti D, Moretto M, Perazzolli M, Si-Ammour A, Vezzulli S, Zini E, Eldredge G, Fitzgerald LM, Gutin N, Lanchbury J, Macalma T, Mitchell JT, Reid J, Wardell B, Kodira C, Chen Z, Desany B, Niazi F, Palmer M, Koepke T, Jiwan D, Schaeffer S, Krishnan V, Wu C, Chu VT, King ST, Vick J, Tao Q, Mraz A, Stormo A, Stormo K, Bogden R, Ederle D, Stella A, Vecchietti A, Kater MM, Masiero S, Lasserre P, Lespinasse Y, Allan AC, Bus V, Chagné D, Crowhurst RN, Gleave AP, Lavezzo E, Fawcett JA, Proost S, Rouzé P, Sterck L, Toppo S, Lazzari B, Hellens RP, Durel CE, Gutin A, Bumgarner RE, Gardiner SE, Skolnick M, Egholm M, Van de Peer Y, Salamini F, Viola R (2010) The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet 42:833–839
Verweij W, Spelt C, Sansebastiano G-P D, Vermeer J, Reale L, Ferranti F, Koes R, Quattrocchio F (2008) An H+ P-ATPase on the tonoplast determines vacuolar pH and flower colour. Nat Cell Biol 10:1456–1462
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wen T, Qing EX, Zeng W, Liu Y (2001) Study on the change of organic acid synthetase activity during fruit development of navel orange (Citrus sinensis Osbeck). J Sichuan Agric Univ 19:27–30
Wu J, Gao H, Zhao L, Liao X, Chen F, Wang Z, Hu X (2007) Chemical compositional characterization of some apple cultivars. Food Chem 103:88–93
Wu J, Li LT, Li M, Khan MA, Li XG, Chen H, Yin H, Zhang SL (2014) High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Exp Bot 65:5771–5781
Xu M, Korban SS (2002) A cluster of four receptor-like genes resides in the Vf locus that confers resistance to apple scab disease. Genetics 162:1995–2006
Xu K, Wang A, Brown S (2012) Genetic characterization of the Ma locus with pH and titratable acidity in apple. Mol Breed 30:899–912
Yao YX, Li M, Zhai H, You CX, Hao YJ (2011) Isolation and characterization of an apple cytosolic malate dehydrogenase gene reveal its function in malate synthesis. J Plant Physiol 168:474–480
Young TE, Juvik JA, Sullivan JG (1993) Accumulation of the components of total solids in ripening fruits of tomato. J Am Soc Hortic Sci 118:286–292
Zhang Q, Ma B, Li H, Chang Y, Han Y, Li J, Wei G, Zhao S, Khan MA, Zhou Y, Gu C, Zhang X, Han Z, Korban SS, Li S, Han Y (2012) Identification, characterization, and utilization of genome-wide simple sequence repeats to identify a QTL for acidity in apple. BMC Genomics 13:537
Zohary D, Hopf M (2000) Domestication of plants in the old word: the origin and spread of cultivated plants in West Asia, Europe and the Nile Valley. Oxford Univ Press, Oxford
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31420103914 and 31372048), the National Basic Research Program of China (Grant No. 2011CB100600), and the National High Technology Research and Development Program of China (Grant No. 2011AA0020401).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by M. Troggio
Data archiving statement
The consensus map of the JG×WSH mapping population is currently being prepared for submitting to Genome Database for Rosaceae (https://www.rosaceae.org).
Rights and permissions
About this article

Cite this article
Ma, B., Zhao, S., Wu, B. et al. Construction of a high density linkage map and its application in the identification of QTLs for soluble sugar and organic acid components in apple. Tree Genetics & Genomes 12, 1 (2016). https://doi.org/10.1007/s11295-015-0959-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-015-0959-6