
Abstract
Brown rot (BR) caused by Monilinia spp. leads to significant post-harvest losses in stone fruit production, especially peach. Previous genetic analyses in peach progenies suggested that BR resistance segregates as a quantitative trait. In order to uncover genomic regions associated with this trait and identify molecular markers for assisted selection (MAS) in peach, an F1 progeny from the cross “Contender” (C, resistant) × “Elegant Lady” (EL, susceptible) was chosen for quantitative trait loci (QTL) analysis. Over two phenotyping seasons, skin (SK) and flesh (FL) artificial infections were performed on fruits using a Monilinia fructigena isolate. For each treatment, infection frequency (if) and average rot diameter (rd) were scored. Significant seasonal and intertrait correlations were found. Maturity date (MD) was significantly correlated with disease impact. Sixty-three simple sequence repeats (SSRs) plus 26 single-nucleotide polymorphism (SNP) markers were used to genotype the C × EL population and to construct a linkage map. C × EL map included the eight Prunus linkage groups (LG), spanning 572.92 cM, with an average interval distance of 6.9 cM, covering 78.73 % of the peach genome (V1.0). Multiple QTL mapping analysis including MD trait as covariate uncovered three genomic regions associated with BR resistance in the two phenotyping seasons: one containing QTLs for SK resistance traits near M1a (LG C × EL-2, R 2 = 13.1–31.5 %) and EPPISF032 (LG C × EL-4, R 2 = 11–14 %) and the others containing QTLs for FL resistance, near markers SNP_IGA_320761 and SNP_IGA_321601 (LG3, R 2 = 3.0–11.0 %). These results suggest that in the C × EL F1 progeny, skin resistance to fungal penetration and flesh resistance to rot spread are distinguishable mechanisms constituting BR resistance trait, associated with different genomic regions. Discovered QTLs and their associated markers could assist selection of new cultivars with enhanced resistance to Monilinia spp. in fruit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brown rot (BR) is a major stone fruit disease, caused by Monilinia spp. necrotrophic fungi (Ascomycota). BR affects stone fruit crops in warm and humid climates worldwide, leading to dramatic productivity losses in the field and in post-harvest operations (EPPO/CABI 1997). Predominant species are Monilinia fructicola in America and Monilinia laxa and Monilinia fructigena in Europe, the latter associated also to BR of pome fruit. Asexual spores or conidia are the major inoculum source of the disease. In immature fruits, conidia can form appressoria to remain quiescent (Adaskaveg et al. 2000; Lee and Bostock 2006, 2007). Hyphae infect fruits by either degrading the cuticle and epidermal tissue (Bostock et al. 1999) or directly entering through pre-existing skin micro-cracks. The fruit is finally rotten and mummified, producing large quantities of spores that can hover through winter and infect plant tissues the following year (Adaskaveg et al. 2008).
Currently, the preferred method to control the disease is the application of fungicides on flowers and fruits. The quantity of applied chemicals can be very high when climatic conditions favor the infection (Yoshimura et al. 2004). For this reason, BR is considered one of the major causes of pesticide use on stone fruits (Ritchie 2000). On the other hand, new strains of Monilinia spp. resistant to some fungicides have been recently reported (Yoshimura et al. 2004; Luo et al. 2008; Amiri et al. 2009, 2010), indicating that such control methods may become poorly effective. An attractive strategy to overcome this problem is the use of cultivars with enhanced disease resistance (Juroszek and Tiedemann 2011), allowing for reduced fungicide inputs and more sustainable and environment-friendly cropping practices.
Breeding programs aimed at enhancing BR resistance are impaired by time-consuming procedures for assessing this trait on field-grown segregating trees. Therefore, an important objective is the generation of new tools for the early selection of seedlings with enhanced BR resistance. Marker-assisted selection (MAS) is a valuable strategy for these purposes, as it allows the early selection of seedlings bearing favorable alleles at marker loci genetically linked to genomic regions that control the trait of interest. In Prunus, the discovery of such molecular markers has been achieved through quantitative trait locus (QTL) mapping on populations derived from biparental crosses. For example, molecular markers have been associated with heritable fruit quality (Quilot et al. 2004; Dirlewanger et al. 2007; Zhang et al. 2009; Eduardo et al. 2010b; Eduardo et al. 2012) and disease resistance traits (Foulongne et al. 2003; Dirlewanger et al. 2004a; Blenda et al. 2007; Marandel et al. 2009). The availability of thousands of molecular markers (Jung et al. 2008) has allowed the construction of multiple genetic maps and the development of synteny studies (Dirlewanger et al. 2004b; Illa et al. 2009). Moreover, the peach genome sequence (Arus et al. 2012;Verde et al. 2013) and re-sequencing of many Prunus cultivars have led to the generation of thousands of single-nucleotide polymorphism (SNP) markers (Sosinski et al. 2009; Ahmad et al. 2011; Verde et al. 2012), whose efficiency is being tested and validated in marker-assisted breeding programs.
Genetic analyses for marker discovery require accurate phenotypic data for the estimation of genotype-associated variation of the trait. However, BR phenotyping is often hindered by environmental and seasonal conditions. For instance, low temperatures (Phillips 1984; Tian and Bertolini 1999), humidity duration (Wilcox 1989; Tamm et al. 1995; Measham et al. 2009), soil nutrient availability (Daane et al. 1995; May-De-Mio et al. 2008), and orchard hygienic conditions (Hong et al. 1997) are factors that significantly influence BR development. Despite these difficulties, cultivar-dependent BR resistance in peach and apricot has been previously evaluated with diverse approaches (Pascal et al. 1994; Bassi et al. 1998; Gradziel et al. 1998; Walter et al. 2004; Wagner Júnior et al. 2008), indicating a quantitative inheritance for this trait and the possibility for its genetic improvement in Prunus. An example is the high BR resistance in the clingstone cultivar “Bolinha,” associated with a more compact arrangement of epidermal and subepidermal cells, thicker cuticle, fewer trichomes, and higher phenolic content at full ripening (Feliciano et al. 1987; Adaskaveg et al. 1991). Despite poor fruit quality (high susceptibility to enzymatic browning, reduced fruit size, and high rate of pre-harvest fruit drop), Bolinha has been used as donor of BR resistance in conventional breeding for canning and low-chill peach (Gradziel et al. 2003; Topp et al. 2008; Wagner Júnior et al. 2008; Wagner Junior et al. 2011). The F1 population from “Contender” × “Elegant Lady” (C × EL) melting flesh peach cross provides an attractive system for the genetic dissection of the trait in cultivars for the fresh market (Bassi et al. 1998; Bassi and Rizzo 2003). Broad-sense heritability has been estimated in this population, with a value of 0.55, although this estimation was biased by the maturity date effect (Pacheco and Bassi 2010).
Factors affecting BR resistance have been suggested in the literature. Skin and cuticle micro-cracking susceptibility is a genetic factor directly associated with BR infection, as it facilitates fungal invasion of the fruit (Borve et al. 2000; Measham et al. 2009). In a previous study (Pascal et al. 1994), skin- and flesh-associated resistance were not correlated in diverse stone fruit species. Biochemical effectors related to BR resistance have been described in stone fruits, such as pathogenesis-related proteins PR-5 and PR-10 in European plum (Prunus domestica; El-kereamy et al. 2009; El-Kereamy et al. 2011) and the antifungal activities of caffeic acid and chlorogenic acid on peach (Bostock et al. 1999; Lee and Bostock 2007; Villarino et al. 2011). Recently, Martinez-Garcia et al. (2013a) reported a first study to associate specific regions of Prunus genome with BR resistance phenotype by QTL analysis, with crosses between peach canning, non-melting cultivars, and peach × almond interspecific hybrids. In the study, the authors identified a wide region on chromosome 1, strongly associated with BR resistance and containing several candidate genes associated with pathogen resistance. However, studies aimed to explore the genomic regions associated with BR resistance existing in other genetic resources, such as melting fleshed peach cultivars, have not been performed to date.
The aim of this work was to identify molecular markers associated with BR resistance in melting fleshed peaches, to gain additional insights into the genetic basis of this complex trait. To this end, a linkage map based on simple sequence repeat (SSR) and SNP markers was constructed for the C × EL F1 population, and QTL analyses were performed with phenotypic data from 2 years of artificial inoculation experiments. The results provide a starting point for future applications of MAS in peach seedlings with enhanced resistance to Monilinia spp., from cultivars for fresh fruit market.
Materials and methods
Plant material
Trees of Contender (C) and Elegant Lady (EL) and 110 seedlings of an F1 population derived from their cross (hereon C × EL) were studied in the years 2009 and 2010. Seedlings were grafted on the GF305 rootstock and planted in 2005 at 1 m × 4 m with two replicate trees per seedling at the experimental orchards of the University of Milan (Azienda Francesco Dotti, Lodi, Italy). Standard cultural practices were carried out, except fungicide applications after petal fall, in order not to add external resistance factors to fruits. Thinning was done before pit hardening, leaving 40–80 fruits according to plant vigor, in order to allow fruit size expression not be limited by competition. Contender (moderately resistant to BR) and Elegant Lady (very susceptible to BR) ripen by the end of July and the first week of August, respectively; the F1 offspring segregates for maturity date (MD) (spanning a period from the first week of July to mid-August), fruit BR resistance, soluble solid content (SSC), and size (Bassi and Rizzo 2003).
Inoculum
M. fructigena strain “Arc1” used in this study was obtained from rotten fruits present in the same orchard of the C × EL progeny. A single colony was stored on potato dextrose agar plates at 4 °C. Molecular analysis according to Hughes et al. (2000) suggested that the isolate corresponded to M. fructigena (not shown). Mycelial plugs were used to infect previously disinfected peach fruits, in order to generate abundant spores for artificial inoculations. After 7 days at 25 °C, spores were washed off from rotten fruits with sterile aqueous 0.05 % Tween 20, and the obtained suspension was filtered in a double layer of sterile cheesecloth. Inoculum concentration was measured on a Malassez counting chamber and adjusted to 5 × 106 spores/ml with sterile aqueous 0.05 % Tween 20.
Infection procedure, phenotyping, and data analysis
In order to minimize BR resistance variation associated with fruit developmental stage, harvest date was determined by the I AD index (Ziosi et al. 2008), not taken invasively in the field with the ∆Ameter instrument (Synteleia S.R.L., Italy). From previous experience, the I AD value for physiological maturity was set to 0.4. I AD was monitored every 2 days by measuring the I AD value in five fruits per seedling, and 40–60 intact and healthy fruits per seedling (according to fruit availability) were harvested when the average value of I AD reached 0.4.
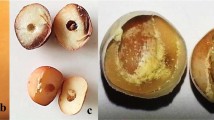
The inoculation procedure was based on published protocols (Pascal et al. 1994; Gradziel et al. 1998; Walter et al. 2004). Fruits of each seedling were then randomly separated in three boxes, each containing 10 to 20 fruits (Electronic Supplementary Material 1). Control treatment (hereon C) was executed by pipetting 10 μl of sterile water over intact and not disinfected fruit skin; preliminary experiments using sterile aqueous 0.05 % Tween 20 in the control treatment showed this to be equivalent to the use of sterile water (data not shown). In order to reduce natural post-harvest fungal infection of treatments including artificial inoculum, harvested fruits were disinfected with 70 % ethanol spray. With the aim to score separately the resistance of skin and flesh, uninjured and artificially injured fruit inoculations were carried out, hereon SK and FL, respectively. SK treatment was performed by applying 10 μl of spore suspension to the surface of the center of the sun-exposed fruit cheek, where fruit cracks are lower than in pedicel and stylar regions (Gibert et al. 2005). FL treatment was accomplished by creating an artificial wound in the center of the fruit, 2 mm deep, with a 1-mm-thick sterile needle and immediately inoculating in the same way as SK. Each fruit box was sealed with plastic wrap to maintain high humidity and was incubated at 25 °C. Five days after inoculation, the diameter of lesions originating from the inoculation point was measured on each fruit with a manual calliper (single rot diameter, hereon “srd,” Electronic Supplementary Material 1). In SK and FL treatments, infections from other points of the fruit were disregarded. In C, as no infections were generated from the inoculation point, any BR infection was considered, in order to take data for natural orchard infections. Percentage of infected fruits (if) and average rot diameter (rd) were considered as susceptibility scores for each seedling under each treatment. Importantly, when infection reached more than half of a given fruit, we considered the rd value as two times the fruit diameter minus the non-rotten diameter.
The measured scores in this work are directly related to disease impact and thus with susceptibility: when we referred to the magnitude of the phenotype, we will use the terms “susceptibility” or “disease impact.” Instead, the term “resistance” refers to a biological phenomenon corresponding to the trait of real interest for breeding, which can be considered as the opposite of susceptibility. Treatment–score combinations considered in this study constituted five susceptibility traits, which are summarized in Table 1.
Statistical analyses including frequency histograms to observe distributions and correlation matrices were performed using R version 2.12.1. Significance of correlations between traits was checked by calculating Spearman’s rank correlation coefficient, given its suitability for not normally distributed data (Foulkes 2009). Susceptibility data were compared between years, treatments, or seedlings by paired Wilcoxon signed-rank and Kruskal–Wallis rank sum tests.
Genotyping
DNA extractions from young leaves of the parents and 110 C × EL seedlings were carried out using the PowerPlant DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA). SSR amplifications were performed following the multiplex-ready PCR protocol (Hayden et al. 2008a; Hayden et al. 2008b) with modifications as in Eduardo et al. (2010a). Allele size was manually determined using the GeneMarker demo version 1.70 (SoftGenetics). Using the multiplex-ready approach with “tagged” SSR primers, the expected size of the alleles corresponded to approximately 30 base pairs larger than reported allele sizes. To search segregating markers on C × EL, 350 Prunus-derived SSRs were screened with the multiplex-ready protocol in the parents and on six test F1 seedlings. Markers giving good peak quality and expected size allele segregation in the test seedlings were then genotyped in the whole C × EL population. The markers to be mapped were also selected on the basis of their position on the available Prunus linkage maps (GDR, http://www.rosaceae.org, “search markers” and “CMap” tool; Jung et al. 2008). To check the position of genotyped markers, we searched their genomic position in the peach genome V1.0 (http://www.rosaceae.org/species/Prunus_persica/genome_v1.0). For markers not present in this database, we estimated their putative genomic positions by aligning primer sequences against the peach genome V1.0 scaffolds database, using batch Blast tool of GDR (http://www.rosaceae.org/bio/content?title=&url=%2Fcgi-bin%2Fgdr%2Fgdr_blast). Marker genomic position was considered as the start of the best match, only if E value <0.005 on both forward and reverse primers. Genotypic data of 26 SNPs from C × EL F1 seedlings genotyped with the Illumina 9,000 SNP array v1 for peach (Verde et al. 2012) was also used in this study. SNP data was available for 80 SSR-genotyped C × EL seedlings. SNPs were selected on the basis of their putative genomic position, in order to fill regions not expected to be covered by the SSR markers (see “Linkage mapping and QTL analysis” section). This SNP data set was kindly provided by Fruit Breedomics consortium through Fondazione Edmund Mach (FEM, San Michele all’Adige, Italy), who performed genotyping reactions as reported by Eduardo et al. (2012).
Linkage mapping and QTL analysis
Genetic linkage analysis and map construction were performed with JoinMap 4.1 (Van Ooijen 2006). In a first map construction step, we used a data set of 80 F1 individuals genotyped with 63 co-dominant SSR markers and 26 SNPs (a total of 89 markers with missing data frequencies lower than 5 %). CP population codes were used for marker grouping. The integrated C × EL map was generated considering simultaneously the maternal and paternal meiosis, by the “Create Population Node” option, since the number of anchor markers found (i.e., markers simultaneously segregating for both parents) allowed this approach. Additionally, this method can deduce the linkage phase and partially estimate recombination frequency of markers with <hk × hk> segregation type. Markers showing segregation distortion were not excluded from the analysis. The pairwise recombination fraction threshold was fixed to 0.4, and recombination frequencies were transformed in genetic distances (centiMorgans, cM) using the Kosambi mapping function. Linkage groups were defined with a minimum logarithm of odds (LOD) value of 5.0 (i.e., the probability that the markers were linked is 105 times higher than the probability they were not), by the independence LOD as grouping test statistic. Linkage groups were constructed using the multipoint maximum likelihood mapping algorithm, using the pre-set default settings as included in the JoinMap 4.1 software (Van Ooijen 2011). Prunus genome scaffolds and C × EL linkage maps were drawn using the MapChart 2.1 software (Voorrips 2002), using marker genomic positions in million base pairs (Mbp) and cM, respectively.
QTL analyses were carried out using the software MAPQTL 6.0 (Van Ooijen 2009). Phenotypic data for MD and the BR resistance traits indicated in Table 1 were collected in 2009 and 2010. In a first analysis, the non-parametric Kruskal–Wallis (KW) rank sum test was used to search phenotype–marker associations without assuming a normal distribution of phenotypic data. In order to obtain an overall significance level of about p = 0.05, a stringent significance level of p = 0.005 was adopted as threshold for the detection of a QTL for the individual test (Van Ooijen 2009). In addition, associations between a trait and a group of linked markers were regarded as robust, even for individual markers with p < 0.05. In order to obtain an estimation of the QTL genomic interval and its contribution to the phenotypic variance, the “interval mapping” (IM) approach was used. After a permutation test for each trait (PT, 10,000 iterations), genome-wide LOD scores corresponding to p = 0.05 were considered as threshold significance levels for the detected QTLs. Given that MD has been reported to be pleiotropic to other quality traits (Eduardo et al. 2010b) and that BR resistance resulted significantly correlated with this trait (see Table 2), the QTL analysis for BR resistance traits was also performed using corresponding season MD data set as covariate (Van Ooijen 2009). For each phenotypic data set (i.e., trait in one season), markers more closely linked to IM-estimated QTL were chosen as candidate co-factors, which were tested with the “automatic cofactor selection” (ACS) tool of MAPQTL 6.0. ACS-confirmed cofactors were used in restricted multiple QTL mapping (rMQM) analysis, in order to absorb residual variance, thus increasing the power for mapping new segregating QTLs. When new segregating QTLs were made evident, new rounds of ACS and rMQM were performed until stabilizing QTL set. MQM was applied in order to resolve the occurrence of multiple QTLs in the same LG. LOD plots of LGs containing QTLs were drawn with the MapChart 2.1 software (Voorrips, 2002). In this work, we consider a QTL as “stable” for a determined trait if it presents a significant effect and is located in similar positions between the two seasons of analysis (2009 and 2010). Also, when a QTL associated with an ACS-validated cofactor marker resulted in a LOD value minor than the PT-determined threshold in 1 year, this QTL was considered as stable if (a) a significant QTL was determined in the same position for the corresponding phenotypic data set from the other year and (a) the difference with LOD threshold was less than 0.5.
Results
Phenotypic data
In this study, a total of 6 traits were analyzed in the C × EL F1 progeny (5 traits shown in Table 1, plus MD) through 2 different seasons, giving place to 12 phenotype data sets. The frequency distributions of these data sets are illustrated in Fig. 1.

Distribution frequency of C × EL F1 progeny for maturity date and BR artificial infection traits in 2 years. Traits and years of analysis are indicated at the top of each histogram. Meanings of trait symbols are indicated in Table 1. Frequency is expressed in the y-axis as the number of individuals falling on each phenotypic interval (x-axis)
All susceptibility traits showed non-normal distributions. Most of the progeny exhibited low infection from field inoculum (C-if trait): infection frequencies lower 10 % were obtained in 36 and 60 seedlings of the progeny in 2009 and 2010, respectively. Regarding SK-rd, distributions were similar among years, showing average infection diameter ranges between 0 and 100 mm and with a major quantity of seedlings with minimum average rot diameters (values minor than 10 mm in around 20 and 50 seedlings in 2009 and 2010, respectively). Around 30 seedlings showed the highest SK-if values in 2009 (more than 90 % of infected fruits), while 30 seedlings showed the lowest rate in 2010 (10 % or less); this difference suggests a strong seasonal effect on this trait. FL-rd showed similar distributions in both years; however, a major number of seedlings with rot diameter values higher than 90 % were registered in 2010. FL-if showed a large number of seedlings with more than 90 % of infected fruits in both years. Contender was almost always more resistant than Elegant Lady. Depending on the trait and score, the parents fell in different infection class in respect to the progeny distributions (Fig. 1). Clear transgressive segregation (i.e., seedlings were observed to have a minor disease impact than Contender parent) was observed for all the analyzed traits (except C-if) on both years of analysis. Thirty-nine transgressive seedlings were observed for SK-rd, 10 for SK-if, 15 for FL-rd, 3 for FL-if, and 23 for MD on the 2 years of phenotypic study (Electronic Supplementary Material 4).
MD showed non-normal distributions in both years. Transgressive segregation for this trait was also observed, since MDs of the parents are close and progeny showed a range of 40–45 days, depending on the year. For some seedlings and for Elegant Lady parent, a delay in MD of 5–10 days was observed from 2009 to 2010, e.g., 10 days delay on Elegant Lady ripening.
Significant differences between all traits were confirmed after performing paired Wilcoxon signed-rank tests (p < 0.0005) confirming a significant effect of the treatment. A lower but significant effect of the year was also detected among all the infection scores (p < 0.05). Kruskal–Wallis rank sum test revealed significant differences between seedlings on both seasons (p < 0.01).
Comparisons of the same traits between seasons resulted in significant correlations for MD, SK-rd, and FL-rd (Table 2a). MD exhibited the highest correlation between years (R 2 = 0.92; p < 0.0005). C-if, SK-if, and FL-if scores gave insignificant correlations between years, indicating a strong seasonal influence in the infection probability at the skin and flesh levels. SK-rd and FL-rd showed R 2 values of 0.29 and 0.39, respectively (p < 0.05), suggesting that they could be controlled by a low but significant genetic component.
Regarding relations between traits (Table 2b), MD significantly correlated with all susceptibility traits (except with C-if in 2010), showing correlations ranging from 0.26 (for SK-if in 2009 with p < 0.05) to 0.72 (for FL-rd in 2009 with p < 0.0005). MD was more correlated (R 2 and significance values) with FL than SK susceptibilities (rd and if), possibly indicating that fruit ripening affects barriers to post-penetration rot spread more than to penetration. C-if correlated only in 2009 with SK-rd and FL-rd (R 2 = 0.34 and 0.28, respectively). Significant correlations were observed between two scores when compared for the same treatment (SK-if vs. SR-rd and FL-if vs. FL-rd); however, R 2 and significance values strongly differed between years. Correlations of 0.55 and 0.47 were observed between SK-rd and FL-rd, in 2009 and 2010, respectively; SK-if and FL-if correlated significantly only in 2010.
Marker transferability and diversity
In order to construct a linkage map for QTL analysis of BR resistance, 350 SSR markers from various Prunus species were selected and tested, identifying 324 that yielded distinguishable peaks in at least one parent (Table 3). The percentage of homozygous and monomorphic markers was 51.7 and 47.7 %, respectively, while 40.8 % of the tested markers resulted heterozygous in at least one parent. Peach SSRs produced higher amplification rate (96.3 vs. 86.3 %), quantity of anchor markers (16.9 vs. 8.3 %), and quantity of heterozygous markers (42.2 vs. 38.6 %), when compared to other Prunus-derived SSRs. Genomic DNA-derived SSRs showed a slightly lower level of amplification (91.9 vs. 94.1 %) compared to EST-SSRs; however, a higher level of heterozigosity (44.7 vs. 31.3 %) and proportion of anchor markers (14.9 vs. 10.7 %) was observed for genomic SSRs.
Ninety-eight and 93 SSR markers were heterozygous in Contender and Elegant Lady, respectively. Eighty-eight SSRs were genotyped in the C × EL population, but only 63 SSRs fulfilling the technical requirements (see “Materials and methods” section) were used for C × EL map construction. Among these, 21 are heterozygous only in Contender, 20 only in Elegant Lady, and 22 on both parents (anchor markers).
In addition, a small set of 26 SNPs genotyped and segregating in the C × EL population were included in the final data set for map construction. Thirteen of these markers are heterozygous only in Contender, 7 only in Elegant Lady, and 6 on both parents.
Linkage analysis
Based on the marker position on the peach genome V1.0, the anchor markers in the population parents were attributed to the eight largest Prunus scaffolds, which currently represent 98.93 % of the genomic sequence (Arus et al. 2012). This allowed the construction of a consensus linkage map for the C × EL F1 (hereon C × EL map; Fig. 2).

Alignment of the C × EL linkage map and peach genome sequence (V1.0). Peach genome scaffolds and C × EL linkage groups are shown on the left and right of each pair, respectively. Chromosomic positions and genetic distances are represented in Mbp (×1.6 scale) and centiMorgan on the same fixed ruler at the left of the figure. Markers included in the C × EL map that were aligned to genome sequence are shown in bold italics. The alignments are represented by solid lines. Heterozygous markers in Contender or Elegant Lady parent are represented with black or white squares, respectively, at the corresponding position on each linkage group. Microsatellites that resulted homozygous in Contender and Elegant Lady are indicated in the genome sequence with the (hm) symbol. Regions of the genome covered by the C × EL map are indicated in gray. Marker skewness significance is represented at the right of each LG (*******p < 10−5; ****p < 5 × 10−3; ***p < 10−2; **p < 5 × 10−2; *p < 10−1). SNP marker names were modified by abbreviating prefixes “SNP_IGA_” and “snp_3_” to “S” and “s3,” respectively
Marker positions in the C × EL linkage map were aligned to the peach genome V1.0 scaffolds, in order to estimate the coverage of C × EL on peach genome. Out of the 383 SSR markers present in this database and used for genome assembly and orientation (Arus et al. 2012), 38 corresponded to SSR included in the C × EL map. The putative position of the remaining markers was estimated in silico (see “Genotyping” section).
The C × EL map covers a total genetic distance of 572.92 cM, corresponding to a physical distance of 171.85 Mbp when aligned to peach genome (78.73 %). The C × EL map consisted in eight linkage groups that covered the eight scaffolds of the peach genome, spanning genetic distances from 54.84 cM (C × EL-8) to 86.55 cM (C × EL-3). The linkage groups that most extensively covered a single scaffold were C × EL-4 and C × EL-7, with 92.16 and 92.99 % of their sequence, respectively. Group C × EL-5 showed the lowest coverage in respect to its corresponding scaffold, with 47.18 %. Single parental maps covered 44.8 and 57.8 % of the C and EL genomes, respectively. Both parental maps covered regions of the eight scaffolds of peach genome and spanned total genetic distances of 439.46 cM in C and 548.21 cM in EL.
The average density of the map was of 6.9 cM/marker. Five events of marker co-segregation were found: one in C × EL-1, where CPPCT034 and UDA-025 co-segregated at 69.20 cM; two in C × EL-4, where ASSR17, ssrPaCITA06, and ssrPaCITA11 co-segregated at 8.21 cM and UDP97-402 and UDAp-439 co-segregated at 49.4 cM; and finally, two in group C × EL-6, where MA040a and UDP98-412 co-localized at 44.72 cM and CPPCT030 and EPPISF002 co-mapped at 57.48 cM. Two gaps larger than 15 cM were found on the C × EL map: on C × EL-1 (16.63 cM from SNP_IGA_10702 to SNP_IGA_19393) and on C × EL-6 (17.62 cM from SNP_IGA_640928 to BPPCT025). No gaps larger than 18 cM were found on C × EL integrated map.
Marker order was conserved between the C × EL map and peach genome V1.0, except for the upper region of C × EL-6, in which the genetic map positions of markers SNP_IGA_615381 and SNP_IGA_604703 (0 and 5.3 cM, respectively) are inverted in respect to their genomic position (3,627,560 and 45,035 bp, respectively). In addition, SNP_IGA_460591, putatively positioned on Scaffold_4, was mapped on C × EL-2.
A strong segregation distortion was found on the upper extreme of C × EL-4, between markers CPSCT039 and SNP_IGA_389048 with the distortion peak on marker CPPCT028 (χ 2 = 37.05 with 3 degrees of freedom (df), p < 10−4). Other regions showing lower levels of segregation distortion were located between markers EPPCU9268 and AMPA103 on group C × EL-4 (χ 2 = 7.20 with 1 df, p < 0.01), between MA069a and BPPCT001 on C × EL-2 (χ 2 = 6.41 with 3 df, p < 0.05), and between CPPCT033 and SNP_IGA_777469 on C × EL-7 (χ 2 = 4.19 with 1 df, p < 0.05). All these markers were included in the analysis since JoinMap 4.1 performs grouping based on the independence LOD score, which is not affected by segregation distortion (Van Ooijen 2006).
QTL analysis
Using MD as covariate in rMQM and MQM analyses, significant marker–phenotype associations with p < 0.05 were found for most of the traits over the 2 years of phenotypic analysis (Table 4). C-if in both years and FL-if in 2010 were exceptions of this, showing no significant QTLs.
Together, we identified a total of 31 statistically significant indications for the presence of QTLs which are located on the eighth LGs of C × EL map (Table 4). Of these indications, 10 were associated to five traits, located in similar positions in the 2 years of observations, and thus considered as stable QTLs in this study. LGs and LOD plots corresponding to these QTLs are shown in Fig. 3.

Location of stable QTLs controlling BR resistance by MQM. Only LGs showing QTLs corresponding to the two seasons under study and in overlapping positions are depicted. Marker names are listed at the right side of each LG and the genetic distances (in centimorgans (cM)) are listed at the left of each one. Parental marker data for the QTL-associated regions is shown at the right of each linkage group, being C1 and C2 the alleles (SSR bands size in base pairs, or SNP variant) of ‘Contender’, and E1 and E2 the alleles of ‘Elegant Lady’. QTLs are drawn at the right of parental marker data of each LG, in the form of LOD plots of MD (left plots, black lines), SK-if (center plots, red lines) and FL-rd (right plots, green lines), in which horizontal axes indicate LOD value for associations of each trait, and vertical axes indicate the genetic positions of the associations on each LG. Vertical dotted lines represent the average value between the genome-wide LOD thresholds corresponding for the two season data sets of each trait. LOD values for 2009 and 2010 data sets are represented with solid and fragmented lines, respectively. Squares inside the lines (solid for 2009 and open for 2010 dataset) indicate marker positions
Two stable QTLs were found for MD. A major QTL, explaining 53.6 and 53.0 % of phenotypic variance in 2009 and 2010, respectively, was found near marker UDP97-402 on group C × EL-4 (MD_2009.4 and MD_2010.4). QTLs were also observed on both years near marker SNP_IGA_604703, on the top of C × EL-6, explaining between 7.9 and 12.5 % of phenotypic variance of MD trait (MD_2009.6 and MD_2010.6, respectively).
SK-rd QTLs were detected for 2009 in LGs C × EL-6 (near marker BPPCT025), C × EL-7 (near UDAp-407, EPPCU5176, and MA061a), and C × EL-8 (near SNP_IGA_874263), explaining between 7.6 and 16.5 % of the phenotypic variation. In 2010, only one QTL was found for SK-rd, linked to marker SNP_IGA_604703 on LG C × EL-6 (R 2 = 20.9 %) and not overlapping with any QTL of 2009 (Table 4).
QTLs associated with SK-if trait were found in C × EL-2, C × EL-4, and C × EL-6 (Table 4 and Fig. 3). We found a stable QTL for this trait in C × EL-2, between markers M1a and UDP96-013 in 2009 (SK-if_2009.2; R 2 = 31.5 %), and near M1a in 2010 (SK-if_2010.2; R 2 = 13.1 %). In C × EL-4, QTLs associated with marker EPPISF032 were observed in both years, SK-if_2009.4 and SK-if_2010.4b, which explained 14.5 and 11.0 % of the phenotypic variance, respectively; in addition, a QTL-linked marker M12a was found only in 2010 (SK-if_2010.4a; R 2 = 16.8 %). In group C × EL-6, a QTL was detected near marker MA040a in 2010 (SK-if_2010.6; R 2 = 13.2 %).
Regarding disease impact on FL treatments, significant QTLs were found in all LGs of C × EL map, except in C × EL-8. For FL-rd treatment, we found minor QTLs for both years in overlapping positions of group C × EL-3, although associated with different co-factor markers (Fig. 3): FL-rd_2009.3 located near SNP_IGA_320761 (R 2 = 3.0 %) and FL-rd_2010.3b associated with co-factor marker SNP_IGA_321601 (R 2 = 11.0 %). In 2009, other minor QTLs for FL-rd were found to locate in C × EL-1 (FL-rd_2009.1 near SNP_IGA_119215, with R 2 = 2.6 %), C × EL-4 (FL-rd_2009.4a near CPPCT028, with R 2 = 2.1 %, and FL-rd_2009.4b near SNP_IGA_401886, with R 2 = 3.0 %), C × EL-5 (FL-rd_2009.5a near SNP_IGA_586225, with R 2 = 2.8 %, and FL-rd_2009.5b near SNP_IGA_598476, with R 2 = 3.5%), C × EL-6 (FL-rd_2009.6 near SNP_IGA_615381, with R 2 = 2.6 %), and C × EL-7 (FL-rd_2009.7 near CPSCT004, with R 2 = 13.0 %). For season 2010, a QTL was found associated with marker EPPCU9268 (FL-rd_2010.4, with R 2 = 23.8 %).
Significant QTLs associated with FL-if trait were found only in 2009 (Table 4), locating in LGs C × EL-1 (FL-if_2009.1 near SNP_IGA_37651, with R 2 = 33.1 %), C × EL-2 (FL-if_2009.2 near BPPCT004, with R 2 = 5.1 %), C × EL-4 (FL-if_2009.4 near SNP_IGA_401886, with R 2 = 13.0 %), and C × EL-5 (FL-if_2009.5 near BPPCT037, with R 2 = 9.7 %).
In order to understand which parent carries favorable alleles for main detected QTLs, genotypic values for SK-if, FL-rd, and MD traits and their associated markers are shown in Table 5.
Regarding SK-if QTLs of the group C × EL-2 (SK-if_2009.2 and SK-if_2010.2), associated markers M1a and UDP96-013 are heterozygous only in Contender parent (lm or 115|111) and homozygous in Elegant Lady (ll or 115|115), thus segregating just in Contender. Interestingly, F1 seedlings carrying lm genotype (C2/E1 and C2/E2 seedlings on Table 5) show higher SK-if scores in both years compared to the ll individuals (C1/E1 and C1/E2). This indicates that in both QTLs SK-if_2009.2 and SK-if_2010.2, the allele C1 (115-bp band) has a positive effect and C2 (111-bp band) has a negative effect for BR resistance. On the other hand, QTLs for SK-if on group C × EL-4 (SK-if_2009.4 and SK-if_2010.4b) are associated with marker EPPISF032, heterozygous on both parents with segregation type <hk × hk> (see SSR alleles on Table 5). From MQM results in the 2 years, C1/E1 and C2/E2 seedlings (hk genotypeor 233|237-bp bands in EPPISF032) showed the highest SK-if values, and C1/E2 and C2/E1 seedlings present the lowest SK-if scores (kk and hh genotypes, or 237|237- and 233|233-bp bands in EPPISF032). C2/E1 scores are higher than C1/E2 in 2009 but similar in 2010. This could be indicating that (a) heterozygosity on this marker has a negative effect on resistance, compared to homozygosity and (b) that C1 allele presents a more positive effect than C2 on BR resistance, more noticeable in 2009 season.
In the case of QTLs FL-rd_2009.3 and FL-rd_2010.3.b, analysis of allele effects is less straightforward because, although their significance interval is overlapping in C × EL-3 (Fig. 3), ACS-selected co-factor markers are not the same and present different segregation types: SNP_IGA_320761 associated to FL-rd_2009.3 is heterozygous in Contender parent only (<lm × ll> segregation type), and SNP_IGA_321601 associated to FL-rd_2010.3b is heterozygous in Elegant Lady only (<nn × np> segregation type). However, as can be noticed from Table 5, C1/E1 seedlings on both FL-rd_2009.3 and FL-rd_2010.3b (having simultaneously AG genotype in SNP_IGA_320761 and AC in SNP_IGA_321601) show the highest levels of FL-rd resistance for the corresponding year; in contrast, C2/E1 (having simultaneously AG genotype in SNP_IGA_320761 and CC in SNP_IGA_321601) seedlings present the lowest levels of BR resistance in both years. This data leads to the hypothesis that allele C2 has a negative effect on BR resistance, in respect to C1.
Regarding MD QTLs, it is shown in Table 5 that major contribution to trait variation is given by segregation of alleles from Contender. For example, in both MD_2009.4 and MD_2010.4 (linked to marker UDP97-402, with <ef × eg> segregation type), a 2-week earliness variation can be observed when comparing seedlings carrying C1 allele in respect to those carrying C2 allele (corresponding to 166 and 167-bp bands in UDP97-402, respectively). Also for QTLs MD_2009.6 and MD_2010.6, a similar trend given by alleles from Contender parent but with a lower phenotypic variation can be noticed.
Discussion
SSR marker genotyping and linkage map
High levels of SSR transferability have been found in rosaceous species (Gasic et al. 2009; Mnejja et al. 2010). In good agreement with previous reports (Eduardo et al. 2010b; Forcada et al. 2012), we successfully amplified 96.3 % of the tested peach SSRs and 86.3 % other Prunus-derived SSRs. The multiplex-ready strategy used for SSR genotyping allowed screening and mapping of a high number of markers (92.5 % of the 350 screened markers, Table 3) with cost- and time-saving efficiency, as previously described in many crop species, including peach, apricot, and cherry (Hayden et al. 2008a; Hayden et al. 2008b; Eduardo et al. 2010b). Observed polymorphism on peach-derived SSRs was higher than for other Prunus-derived SSRs and higher for genomic SSRs than for cDNA SSRs (Table 3), as previously reported in peach (Dirlewanger et al. 2007; Eduardo et al. 2010b) and other Rosaceae (Gasic et al. 2009; Mnejja et al. 2010). Parents exhibited similar proportion of heterozygous markers (28 % for Contender and 26.5 % for Elegant Lady, Table 3) but lower than the 35 % of heterozygosity found previously on a panel of 212 commercial peach cultivars analyzed with 50 SSR markers (Aranzana et al. 2010).
From the 89 markers selected to genotype the C × EL progeny, 28 corresponded to anchor markers (heterozygous in both parents). With this data set and using the population node option and multipoint maximum likelihood mapping strategy included in JoinMap 4.1 (Van Ooijen 2011), a unified map for the C × EL F1 progeny was successfully constructed (Fig. 2). This strategy was chosen also because it allowed us to use markers with <hk × hk> segregation type, representing a high proportion of the anchor markers of the genotype dataset (16 of the 28 anchors). A similar approach has been used in the map construction and genetic analysis of a full-syb F1 mapping population of Miscanthus sinensis (Swaminathan et al. 2012).
Using marker position on the peach genome V1.0, this map was estimated to cover 78.3 % of the genomic sequence. The total genetic distance covered by the C × EL map (572.2 cM) was higher than the distance of the Prunus reference map (T × E, 519 cM; Dirlewanger et al. 2004a), possibly because of the high number of used <hk × hk> anchor markers, which are informative just for the homozygous individuals, not for the heterozygous. This loss of information can result in distortions on the genetic distance estimations. Moreover, the recombination events of both C and EL parents were integrated in a single map. Illa et al. (2009) suggested that crosses involving similar genomes show higher recombination rates than interspecific crosses, as the case of T × E. Other peach intraspecific maps (Yamamoto et al. 2005; Blenda et al. 2007; Dirlewanger et al. 2007) cover more extensive genetic distances, compared with T × E Prunus reference map because of the higher number of markers and individuals used. Improvement of coverage and marker density could be achieved by increasing marker number. For instance, in the “Bolero × OroA” F1 mapping population, the addition of ~270 SNP markers (Eduardo et al. 2012) allowed the coverage of entire linkage groups that were absent in a previous effort (Eduardo et al. 2010b), although other scaffolds remained without coverage. In this work, the addition of 26 SNPs to the SSR data set contributed to cover regions that appeared as extensively homozygous when only genotyped with SSR markers on the parents (markers indicated as “(hm)” on genome scaffolds shown in Fig. 2). For instance, scaffold 3 is not covered by SSRs in Contender genome, as just <nn × np> segregation type markers were found. The integration between SSR and SNP markers resulted in the absence of gaps larger than 20 cM, with the largest gap (17.6 cM) located on the middle of group C × EL-6, one of the genomic regions with extensive homozygosity. Low levels of correlation between SSR and SNP marker heterozygosity have been detected on peach germplasm (Aranzana et al. 2012); this evidence supports the integration of such marker types into single data sets. The lower levels of physical distance of the peach genome coverage of single parental map (44.8 % for C and 57.8 % for EL) have been also observed in other parental maps form F1 populations, such as Bolero and OroA maps (Eduardo et al. 2012), in which some entire groups remained uncovered, as mentioned before, although large initial marker sets were used to map construction. As discussed in that study and in Stange et al. (2013), identity by descent (a consequence of the narrow genetic diversity of the modern peach cultivars; Scorza et al. 1985) can explain the extensive homozygous regions found in parental genomes. In the regions covered by the C × EL map, marker density was 6.9 cM/marker. We regard this map as sufficiently saturated for QTL detection and BR-associated marker discovery, since high levels of conservation of linkage disequilibrium have been observed in peach, extending up to 13–15 cM (Aranzana et al. 2010); this suggests that a map with such density has enough power of detection of QTLs. In addition, simulation experiments on maize genome demonstrated that the increase in map density improves neither the power of detection nor the power of prediction of explained variance of a QTL (Stange et al. 2013).
An inversion was observed in the top of group C × EL-6, between markers SNP_IGA_615381 and SNP_IGA_604703; also, SNP_IGA_460591, expected to be on Scaffold_4, was mapped in C × EL-2. These results could be explained by a misassembly in this region of peach genome (Verde et al. 2013). Similar results have been obtained in other works of peach linkage mapping using SNP markers (Eduardo et al. 2012; Martinez-Garcia et al. 2013b).
Response to BR infection in peach
Fruit epicarp has been extensively indicated as an important broad range resistance source against opportunistic pathogens such as Monilinia spp. In this study, besides considering the presence and absence of this main barrier, two infection parameters were scored after direct fungal inoculation on fruit flesh. With this infection approach, seasonal correlations for rot diameter in the presence and absence of skin barrier were observed (SK-rd and FL-rd; Table 2). Considering these correlations, the occurrence of a significant genetic component controlling the resistance to infection expansion after penetration could be suggested. Walter et al. (2004) showed a significant correlation in BR infection area between wounded and not wounded apricots (47 % with p < 0.01). These results were in contrast with previous studies (Pascal et al. 1994), where no correlation was found between skin and flesh infection on fruits of several Prunus cultivars. Walter et al. (2004) argued that discrepancy in the results was due to technical differences in the inoculation procedures. In addition, the “low correlation” mentioned in Pascal et al. (1994) referred only to the lack of similarity between the rankings of cultivar disease impact obtained after skin and flesh infections and not a regression-based correlation between two treatments.
Low seasonal correlations for SK-if were found in this study (high levels of infection in 2009 and low levels in 2010; Fig. 1), as well as C-if trait. SK assays were designed in order to represent infection of BR on fruit skin occurring in uniform conditions of humidity, temperature, fruit ripening stage, and inoculum quantity and strain. The observed variation across years of phenotyping can be due to environmental factors influencing fruit barrier efficiency (Gibert et al. 2005; Gibert et al. 2009; Measham et al. 2009). Since C-if infections are the result of the presence of natural inoculum on the studied fruits, in this treatment, environmental variability is also influencing fungal pathogenicity (Tian and Bertolini 1999), and thus, low seasonal correlations should be expected. Stabilization of climatic factors and inoculum presence could be achieved by greenhouse tree growing; however, this strategy is not cost-effective and it does not reflect true conditions of peach production. No correlations were detected between C-if and SK-if in any year (Table 2); however, the trend in overall infection levels is similar since C-if and SK-if values were higher in 2009 than in 2010, being C-if seasonal variation less dramatic than in SK-if (Fig. 1). These results could suggest that in two different seasons, environmental factors influencing fruit resistance and fungal pathogenicity resulted in similar trends for two completely different phenotype scores (see Table 1 and “Materials and methods” section).
In our opinion, and considering the genetic background analyzed in this study, the most informative treatment in terms of BR resistance QTLs discovery was SK-if. Unwounded inoculations (SK) led to the discovery of two resistance QTLs that showed significant effects in the 2 years of study (C × EL-4 and C × EL-2, see below for further discussion on allelic effects). FL inoculations were important in this study as season-repeated QTLs were identified in LG C × EL-3, with minor but significant effects. However, FL could be helpful in the detection of QTLs for BR expansion resistance in other peach genetic backgrounds, providing new insights in defense mechanisms against necrotrophic pathogens on mature fruits. By contrast, we did not find QTLs with significant effects for C-if trait, probably due to the strong influence of the environment on components of natural infections, such as fruit defenses, fungal aggressiveness, and the interaction between fruit and pathogen (Tian and Bertolini 1999). In conclusion, resistance to penetration is more interesting for a breeder or a grower, since fruits once infected are lost. For these reasons, we recommend concentrating in phenotyping efforts on SK infections.
Maturity date as covariate in MQM analyses
MD showed significant seasonal correlation (92 % with p < 0.0005; Table 2), and strong QTLs were found in C × EL-4, explaining around 50 % of the trait variability (Table 4 and Fig. 3). MD_2009.4 and MD_2010.4 QTLs coincide with the position of a major MD locus previously found in a related progeny (Eduardo et al. 2010b; Pirona et al. 2013), as well as others from Prunus genetic backgrounds (Quilot et al. 2004) and in apple (Kenis et al. 2008), this latter associated with loci in Malus LG3 and LG10, previously described as syntenic with LG4 of Prunus (Dirlewanger et al. 2004b; Illa et al. 2011). In addition to its strong effect in MD phenotype, in this study, MD QTLs of LG4 positively correlated in both years to most of the traits in at least one season (Table 2). The correlation observed between C-if and MD (20 % with p < 0.005) is consistent with the hypothesis that lateness is proportional to pathogen exposure in the orchard, i.e., late-ripening cultivars are more prone to Monilinia spp. infection (Topp et al. 2008). Artificial inoculation traits (SK and FL) are also correlated with MD. This is in agreement with the obtained 9 BR resistance QTLs (out of 12) that clustered in the MD-associated region on LG4, when preliminary IM QTL analysis was performed (Electronic Supplementary Material 2 and 3). This MD locus on LG4 has previously been proposed to exert pleiotropic effects on other fruit traits of Prunus and Malus, such as fruit size, SSC, and acidity (Eduardo et al. 2010b), as well as chilling injury (CI) susceptibility traits, which collocated with a MD QTL in an analogous position of linkage LG4 of a “Venus” × “Big Top” F1 map (Cantin et al. 2010). Ripening is the sum of multiple biochemical and physiological changes that occur at the final stage of fruit development (Giovannoni 2001). BR resistance decreases in stone fruit between pit hardening and physiological maturity (Biggs and Northover 1988; Mari et al. 2003). This change has been suggested to be due to a concomitant drop in the fruit phenolic content (Lee and Bostock 2006, 2007; Villarino et al. 2011). Based on our’s and previous results, the relationship between MD and flesh BR resistance in C × EL may be linked to varying decay of fruit barriers during the ripening process.
These data support the validity of MD trait as a covariate factor of BR resistance in fruits, leading to a more precise genetic dissection of this trait. After applying MD as covariate in an MQM analysis, most of the BR resistance QTLs in the IM analyses were related to C × EL-4 (Electronic Supplementary Material 2 and 3) and disappeared or fell far below the LOD significance threshold; conversely, 27 significant BR resistance QTLs became evident in 23 different loci that distributed in the 8 covered LGs (Fig. 3 and Table 4). In the particular case of SK-if QTLs of group C × EL-4, the MQM analyses with MD covariate allowed to resolve in a precise way the association of this trait with markers EPPISF032 (seasons 2009 and 2010) and M12a (season 2010) and not with MD QTL, as resulted after IM analysis. Selection of significant covariates as factors for genetic models such as MQM mapping has been a successfully used strategy in precise QTL mapping of other traits of agricultural interest in different crops, such as waterlogging tolerance in barley, in which salinity tolerance was used as a covariate (Zhou et al. 2012). Similarly, De Koeyer et al. (2004) determined that the most accurate models to map QTLs for grain quality traits in spring oat were those including covered/hulless trait as a covariate, as it significantly correlated with traits of interest.
QTLs for resistance to BR infection
SK traits reflect the resistance to fungal penetration into the fruit exocarp, a strong physical and chemical barrier. Detection of season-repeated QTLs for SK-if on C × EL-2 and C × EL-4 by MQM QTL analysis including MD as covariate suggests that resistance components independent from MD are being expressed differentially among C × EL seedlings (Tables 4 and 5, Fig. 3). Few disease resistance QTLs have been mapped in the region associated with SK-if QTLs of C × EL-2. In the “Prunus resistance map” (Lalli et al. 2005), two resistance gene analogues (RGAs) have been anchored to LG2. One of these RGAs is situated in the lower region of T × E-2 group (marker AC19, 10 cM under UDP96-013), collocating with a QTL for powdery mildew resistance (Foulongne et al. 2003). QTLs for bacterial spot of leaf and fruit tissues of peach have been recently reported (Yang et al. 2013), indicating the presence of several putative R genes on the detected QTL regions. However, these RGAs encode nucleotide binding site leucine-rich repeat (NBS-LRR)-type proteins, which have been related predominantly to the regulation of resistance responses to biotrophic pathogens (such as powdery mildew and Xanthomonas spp. bacterial spot), leading to a hypersensitive response (HR), a host-induced cell death at the site of infection blocking pathogen expansion via nutrient limitation (Jones and Dangl 2006; Mengiste 2012). Such cell death in contrast can be beneficial for necrotrophics and can also induce host cell death (Govrin et al. 2006). About QTLs associated with marker EPPISF032 in C × EL-4, like QTLs of C × EL-2, few resistance loci can be found in literature. Markers linked with ESTs related with resistance to powdery mildew (a biotrophic pathogen) were found in Lalli et al. (2005) in a similar map position of LG4 (markers CC136A, AG8A, and CC138, positioned near BPPCT015 and thus to EPPISF032). A QTL was recently found in Martinez-Garcia et al. (2013a) in LG4 of “PopDF” (see below); however, the QTL resulted significant only 1 year, and significance interval is in a position around 8,500,000 bp of scaffold 4, far from the position of QTLs SK-if_2009.4 and SK-if_2010.4b (near EPPISF032, around 12.77 Mb of the same scaffold).
Recently, QTL analysis for BR resistance in peach has been published (Martinez-Garcia et al. 2013a). In the cited work, the authors found two candidate QTLs on the LG1, significantly associated with skin BR resistance in 2 out of 3 years of phenotypic analysis. Although there is a similarity between the present work and that of Martinez-Garcia et al. (2013a) in terms of experimental approach, these works differ in fundamental points that have led to different results. First, the genetic backgrounds of the analyzed populations in the cited and the present study are different: in Martinez-Garcia et al. (2013a), the studied population is PopDF (cross “Dr. Davis” × “F8,1-42”), with pedigrees including mostly historical canning peach cultivars and almond introgressions; by contrast, most accessions constituting the pedigrees of Contender and Elegant Lady (parents of the population used in the present study) correspond to traditional, melting fleshed peach cultivars, characterized by a recurrent presence of “Chinese Cling” and “JH Hale,” and any introgression from other Prunus species is observed in their lineages. In the case of quantitative traits, like BR resistance, favorable alleles segregating in a population can be absent in populations with a different genetic background, carrying to the discovery of different QTLs, depending on the population studied. An example of this can be found in Eduardo et al. (2010b), where different QTL MD traits were found in different genomic chromosomes depending the population analyzed. Another key difference between these studies is the statistical approach used to discover QTLs: in the latter, the authors show “candidate QTLs” determined with IM of MapQTL software; in our research instead, rMQM analysis was applied and presented QTLs have been determined after a complete analysis. IM is an initial step in QTL analysis, followed by the use of co-factors in a MQM or rMQM, as indicated by software authors (Van Ooijen 2009). MQM and rMQM are implemented to use co-factors (i.e., markers near candidate QTLs determined by IM and confirmed by ACS) in a multiple QTL model that results in a substantial reduction of the residual variance, increasing the QTL analysis power; also, with the use of co-factors, QTL effects are adjusted for unequal presence of additional QTLs on both allelic classes of a parental locus, which can increase or decrease the significance of the QTL. This approach has been performed on QTL analysis for different traits and plant species (Singh et al. 2009; Rant et al. 2013).
From the presented results of F1 seedling genotypic values (Table 5), it can be noticed that mainly Contender parent transmits variation in QTLs for SK-if of C × EL-2, being the presence of C1 allele of SK-if_2009.2 and SK-if_2010.2 (in phase with 115-bp band in marker M1a) associated with more resistant phenotypes. In addition, in respect to QTLs of group C × EL-4 (linked to EPPISF032), alleles conferring skin resistance come from both parents, as more resistant seedlings have C1/E2 and C2/E1 QTL genotypes). These findings indicate that Contender and Elegant Lady are carrying alleles on at least two genomic regions with both negative and positive effects to the probability of active penetration by M. fructigena. This could explain the presence of transgressive seedlings in almost all traits considered in this study (see Supplementary Material 4). Especially for SK-if, a high proportion of seedlings classified as transgressive in both years of analysis (7 out of 10) simultaneously carried favorable alleles of M1a and EPPISF032.
Concluding remarks
QTLs identified in this work point to few genomic regions underlying BR response traits in the C × EL cross and provide a starting point for the development of MAS for increased resistance to BR. However, further dissection of these chromosomal regions is required to obtain tightly linked markers, and F2 populations need to be studied to evaluate with more precision the additive, dominance effects and/or epistatic interactions of these QTLs, as well as to detect new regions contributing to the phenotypic variation on the studied populations. The recent publication of the peach genome (Verde et al. 2013) and the previously released SNP platforms, allowing simultaneous analysis in thousands of SNPs (Ahmad et al. 2011; Verde et al. 2012), offer unprecedented opportunities to find polymorphic markers in order to saturate these regions.
References
Adaskaveg JE, Feliciano AJ, Ogawa JM (1991) Evaluation of the cuticle as a barrier to penetration by Monilinia fructicola. Phytopathology 81(10):1150
Adaskaveg JE, Forster H, Thompson DF (2000) Identification and etiology of visible quiescent infections of Monilinia fructicola and Botrytis cinerea in sweet cherry fruit. Plant Dis 84(3):328–333
Adaskaveg JE, Schnabel G, Förster H (2008) Diseases of peach caused by fungi and fungal-like organisms: biology, epidemiology and management. In: Layne DR, Bassi D (eds) The Peach: Botany, Production and Uses. CABI, Wallingford, UK
Ahmad R, Parfitt DE, Fass J, Ogundiwin E, Dhingra A, Gradziel TM, Lin D, Joshi NA, Martinez-Garcia PJ, Crisosto CH (2011) Whole genome sequencing of peach (Prunus persica L.) for SNP identification and selection. BMC Genomics 12:569
Amiri A, Brannen PM, Schnabel G (2009) Resistance to respiration inhibitor fungicides in Monilinia fructicola field isolates from South Carolina and Georgia. Phytopathology 99(6):S3–S3
Amiri A, Brannen PM, Schnabel G (2010) Reduced sensitivity in monilinia fructicola field isolates from south carolina and georgia to respiration inhibitor fungicides. Plant Dis 94(6):737–743. doi:10.1094/pdis-94-6-0737
Aranzana MJ, Garcia-Mas J, Carbo J, Arus P (2002) Development and variability analysis of microsatellite markers in peach. Plant Breed 121(1):87–92
Aranzana M, Abbassi E-K, Howad W, Arus P (2010) Genetic variation, population structure and linkage disequilibrium in peach commercial varieties. BMC Genet 11(1):69. doi:10.1186/1471-2156-11-69
Aranzana MJ, Illa E, Howad W, Arus P (2012) A first insight into peach Prunus persica (L.) Batsch SNP variability. Tree Genet Genome 8(6):1359–1369. doi:10.1007/s11295-012-0523-6
Arus P, Verde I, Sosinski B, Zhebentyayeva T, Abbott AG (2012) The peach genome. Tree Genet Genome 8(3):531–547. doi:10.1007/s11295-012-0493-8
Bassi D, Rizzo M (2003) Breeding peaches for fruit quality and brown-rot [(Monilia laxa Aderh. et Ruhl. (Honey)] resistance in peach [Prunus persica (L.) Batsch - Emilia-Romagna]. Italus Hortus 10(5):60–65
Bassi D, Rizzo M, Cantoni L (1998) Assaying brown rot [(Monilinia laxa aderh. et ruhl. (honey)] susceptibility in peach cultivars and progeny. Acta Horticult (ISHS) 465:715–722
Biggs AR, Northover J (1988) Early and late-season susceptibility of peach fruits to monilinia fructicola. Plant Dis 72(12):1070–1074
Blenda AV, Verde I, Georgi LL, Reighard GL, Forrest SD, Munoz-Torres M, Baird WV, Abbott AG (2007) Construction of a genetic linkage map and identification of molecular markers in peach rootstocks for response to peach tree short life syndrome. Tree Genet Genome 3(4):341–350. doi:10.1007/s11295-006-0074-9
Borve J, Sekse L, Stensvand A (2000) Cuticular fractures promote postharvest fruit rot in sweet cherries. Plant Dis 84(11):1180–1184
Bostock RM, Wilcox SM, Wang G, Adaskaveg JE (1999) Suppression of Monilinia fructicola cutinase production by peach fruit surface phenolic acids. Physiol Mol Plant Pathol 54(1–2):37–50
Cantin CM, Crisosto CH, Ogundiwin EA, Gradziel T, Torrents J, Moreno MA, Gogorcena Y (2010) Chilling injury susceptibility in an intra-specific peach Prunus persica (L.) Batsch progeny. Postharvest Biol Technol 58(2):79–87. doi:10.1016/j.postharvbio.2010.06.002
Cantini C, Iezzoni AF, Lamboy WF, Boritzki M, Struss D (2001) DNA fingerprinting of tetraploid cherry germplasm using simple sequence repeats. J Am Soc Hortic Sci 126(2):205–209
Cipriani G, Lot G, Huang WG, Marrazzo MT, Peterlunger E, Testolin R (1999) AC/GT and AG/CT microsatellite repeats in peach Prunus persica (L) Batsch : isolation, characterisation and cross-species amplification in Prunus. Theor Appl Genet 99(1–2):65–72
Clarke JB, Tobutt KR (2003) Development and characterization of polymorphic microsatellites from Prunus avium‘Napoleon’. Mol Ecol Notes 3(4):578–580. doi:10.1046/j.1471-8286.2003.00517.x
Clarke JB, Sargent DJ, Bošković RI, Belaj A, Tobutt KR (2008) A cherry map from the inter-specific cross Prunus avium ‘Napoleon’× P. nipponica based on microsatellite, gene-specific and isoenzyme markers. Tree Genet Genome 5(1):41–51. doi:10.1007/s11295-008-0166-9
Daane KM, Johnson RS, Michailides TJ, Crisosto CH, Dlott JW, Ramirez HT, Yokota GY, Morgan DP (1995) Excess nitrogen raises nectarine susceptibility to disease and insects. Calif Agric 49(4):13–18. doi:10.3733/ca.v049n04p13
De Koeyer DL, Tinker NA, Wight CP, Deyl J, Burrows VD, O’Donoughue LS, Lybaert A, Molnar SJ, Armstrong KC, Fedak G, Wesenberg DM, Rossnagel BG, McElroy AR (2004) A molecular linkage map with associated QTLs from a hulless x covered spring oat population. Theor Appl Genet 108(7):1285–1298. doi:10.1007/s00122-003-1556-x
Dirlewanger E, Cosson P, Tavaud M, Aranzana MJ, Poizat C, Zanetto A, Arus P, Laigret F (2002) Development of microsatellite markers in peach Prunus persica (L.) Batsch and their use in genetic diversity analysis in peach and sweet cherry (Prunus avium L.). Theor Appl Genet 105(1):127–138. doi:10.1007/s00122-002-0867-7
Dirlewanger E, Cosson P, Howad W, Capdeville G, Bosselut N, Claverie M, Voisin R, Poizat C, Lafargue B, Baron O, Laigret F, Kleinhentz M, Arus P, Esmenjaud D (2004a) Microsatellite genetic linkage maps of myrobalan plum and an almond-peach hybrid - location of root-knot nematode resistance genes. Theor Appl Genet 109(4):827–838. doi:10.1007/s00122-004-1694-9
Dirlewanger E, Graziano E, Joobeur T, Garriga-Caldere F, Cosson P, Howad W, Arus P (2004b) Comparative mapping and marker-assisted selection in Rosaceae fruit crops. Proc Natl Acad Sci U S A 101(26):9891–9896. doi:10.1073/pnas.0307937101
Dirlewanger E, Cosson P, Boudehri K, Renaud C, Capdeville G, Tauzin Y, Laigret F, Moing A (2007) Development of a second-generation genetic linkage map for peach Prunus persica (L.) Batsch and characterization of morphological traits affecting flower and fruit. Tree Genet Genome 3(1):1–13. doi:10.1007/s11295-006-0053-1
Downey SL, Iezzoni AF (2000) Polymorphic DNA markers in black cherry (Prunus serotina) are identified using sequences from sweet cherry, peach, and sour cherry. J Am Soc Hortic Sci 125(1):76–80
Eduardo I, Chietera G, Bassi D, Rossini L, Vecchietti A (2010a) Identification of key odor volatile compounds in the essential oil of nine peach accessions. J Sci Food Agric 90(7):1146–1154. doi:10.1002/jsfa.3932
Eduardo I, Pacheco I, Chietera G, Bassi D, Pozzi C, Vecchietti A, Rossini L (2010b) QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genome 7(2):323–335. doi:10.1007/s11295-010-0334-6
Eduardo I, Chietera G, Pirona R, Pacheco I, Troggio M, Banchi E, Bassi D, Rossini L, Vecchietti A, Pozzi C (2012) Genetic dissection of aroma volatile compounds from the essential oil of peach fruit: QTL analysis and identification of candidate genes using dense SNP maps. Tree Genet Genome. doi:10.1007/s11295-012-0546-z
El-kereamy A, Jayasankar S, Taheri A, Errampalli D, Paliyath G (2009) Expression analysis of a plum pathogenesis related 10 (PR10) protein during brown rot infection. Plant Cell Rep 28(1):95–102. doi:10.1007/s00299-008-0612-z
El-Kereamy A, El-Sharkawy I, Ramamoorthy R, Taheri A, Errampalli D, Kumar P, Jayasankar S (2011) Prunus domestica pathogenesis-related protein-5 activates the defense response pathway and enhances the resistance to fungal infection. Plos One 6 (3). doi:e1797310.1371/journal.pone.0017973
EPPO/CABI (1997) Data sheets on quarantine organisms No. 153, Monilinia fructicola. In: Smith IMMD, Scott PR, Holderness M (eds) Quarantine pests for Europe, 2nd edn. CABI International, Wallingford, UK
Feliciano A, Feliciano AJ, Ogawa JM (1987) Monilinia fructicola resistance in the peach cultivar ‘Bolinha’. Phytopathology 77(6):776–780. doi:10.1094/Phyto-77-776
Forcada CFI, Marti AFI, Company RSI (2012) Mapping quantitative trait loci for kernel composition in almond. BMC Genet 13:9. doi:10.1186/1471-2156-13-47
Foulkes AS (2009) Applied statistical genetics with r: for population-based association studies. Use R! (c) Springer Science+Business MEdia, New York. doi:10.1007/978-0-387-89554-3_1
Foulongne M, Pascal T, Pfeiffer F, Kervella J (2003) QTLs for powdery mildew resistance in peach x Prunus davidiana crosses: consistency across generations and environments. Mol Breed 12(1):33–50
Gasic K, Han YP, Kertbundit S, Shulaev V, Iezzoni AF, Stover EW, Bell RL, Wisniewski ME, Korban SS (2009) Characteristics and transferability of new apple EST-derived SSRs to other Rosaceae species. Mol Breed 23(3):397–411. doi:10.1007/s11032-008-9243-x
Georgi LL, Wang Y, Yvergniaux D, Ormsbee T, Inigo M, Reighard G, Abbott AG (2002) Construction of a BAC library and its application to the identification of simple sequence repeats in peach Prunus persica (L.) Batsch. Theor Appl Genet 105(8):1151–1158. doi:10.1007/s00122-002-0967-4
Gibert C, Lescourret F, Genard M, Vercambre G, Pastor AP (2005) Modelling the effect of fruit growth on surface conductance to water vapour diffusion. Ann Bot 95(4):673–683. doi:10.1093/aob/mci067
Gibert C, Chadoeuf J, Nicot P, Vercambre G, Genard M, Lescourret F (2009) Modelling the effect of cuticular crack surface area and inoculum density on the probability of nectarine fruit infection by Monilinia laxa. Plant Pathol 58(6):1021–1031. doi:10.1111/j.1365-3059.2009.02121.x
Giovannoni J (2001) Molecular biology of fruit maturation and ripening. Annu Rev Plant Physiol Plant Mol Biol 52:725–749
Govrin EM, Rachmilevitch S, Tiwari BS, Soloman M, Levine A (2006) An elicitor from Botrytis cinerea induces the hypersensitive response in Arabidopsis thaliana and other plants and promotes the gray mold disease. Phytopathology 96(3):299–307. doi:10.1094/phyto-96-0299
Gradziel TM, Thorpe MA, Bostock RM, Wilcox S (1998) Breeding for brown rot (Monilinia fructicola) resistance in clingstone peach with emphasis on the role of fruit phenolics. Acta Horticult (ISHS) 465:161–170
Gradziel TM, Bostock RM, Adaskaveg JE (2003) Resistance to brown rot disease in peach is determined by multiple structural and biochemical components. Acta Horticult 622:347–352
Hagen LS, Chaib J, Fady B, Decroocq V, Bouchet JP, Lambert P, Audergon JM (2004) Genomic and cDNA microsatellites from apricot (Prunus armeniaca L.). Mol Ecol Notes 4(4):742–745
Hayden MJ, Nguyen TM, Waterman A, Chalmers KJ (2008a) Multiplex-Ready PCR: a new method for multiplexed SSR and SNP genotyping. BMC Genomics 9(1):80. doi:10.1186/1471-2164-9-80
Hayden MJ, Nguyen TM, Waterman A, McMichael GL, Chalmers KJ (2008b) Application of multiplex-ready PCR for fluorescence-based SSR genotyping in barley and wheat. Mol Breed 21(3):271–281. doi:10.1007/s11032-007-9127-5
Hong CX, Holtz BA, Morgan DP, Michailides TJ (1997) Significance of thinned fruit as a source of the secondary inoculum of Monilinia fructicola in California nectarine orchards. Plant Dis 81(5):519–524
Howad W, Yamamoto T, Dirlewanger E, Testolin R, Cosson P, Cipriani G, Monforte AJ, Georgi L, Abbott AG, Arus P (2005) Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics 171(3):1305–1309. doi:10.1534/genetics.105.043661
Hughes KJD, Fulton CE, McReynolds D, Lane CR (2000) Development of new PCR primers for identification of Monilinia species. EPPO conference on diagnostic techniques for plant pests, Waddington, Netherlands, 1-4 February 2000 30:507-511
Illa E, Lambert P, Quilot B, Audergon JM, Dirlewanger E, Howad W, Dondini L, Tartarini S, Lain O, Testolin R, Bassi D, Arus P (2009) Linkage map saturation, construction, and comparison in four populations of Prunus. J Horticult Sci Biotechnol:168-175
Illa E, Sargent DJ, Girona EL, Bushakra J, Cestaro A, Crowhurst R, Pindo M, Cabrera A, van der Knaap E, Iezzoni A, Gardiner S, Velasco R, Arus P, Chagne D, Troggio M (2011) Comparative analysis of rosaceous genomes and the reconstruction of a putative ancestral genome for the family. BMC Evol Biol 11. doi:10.1186/1471-2148-11-9
Jones JDG, Dangl JL (2006) The plant immune system. Nature 444(7117):323–329. doi:10.1038/nature05286
Jung S, Staton M, Lee T, Blenda A, Svancara R, Abbott A, Main D (2008) GDR (Genome Database for Rosaceae): integrated web-database for Rosaceae genomics and genetics data. Nucleic Acids Res 36:D1034–D1040. doi:10.1093/nar/gkm803
Juroszek P, Tiedemann A (2011) Potential strategies and future requirements for plant disease management under a changing climate. Plant Pathol 60(1):100–112. doi:10.1111/j.1365-3059.2010.02410.x
Kenis K, Keulemans J, Davey MW (2008) Identification and stability of QTLs for fruit quality traits in apple. Tree Genet Genome 4(4):647–661. doi:10.1007/s11295-008-0140-6
Lalli DA, Decroocq V, Blenda AV, Schurdi-Levraud V, Garay L, Le Gall O, Damsteegt V, Reighard GL, Abbott AG (2005) Identification and mapping of resistance gene analogs (RGAs) in Prunus: a resistance map for Prunus. Theor Appl Genet 111(8):1504–1513. doi:10.1007/s00122-005-0079-z
Lee MH, Bostock RM (2006) Induction, regulation, and role in pathogenesis of appressoria in Monilinia fructicola. Phytopathology 96(10):1072–1080. doi:10.1094/phyto-96-1072
Lee MH, Bostock RM (2007) Fruit exocarp phenols in relation to quiescence and development of Monilinia fructicola infections in Prunus spp.: a role for cellular redox? Phytopathology 97(3):269–277. doi:10.1094/phyto-97-3-0269
Lopes MS, Sefc KM, Laimer M, Machado AD (2002) Identification of microsatellite loci in apricot. Mol Ecol Notes 2(1):24–26
Luo CX, Cox KD, Amiri A, Schnabel G (2008) Occurrence and detection of the DMI resistance-associated genetic element ‘Mona’ in Monilinia fructicola. Plant Dis 92(7):1099–1103. doi:10.1094/pdis-92-7-1099
Marandel G, Salava J, Abbott A, Candresse T, Decroocq V (2009) Quantitative trait loci meta-analysis of Plum pox virus resistance in apricot (Prunus armeniaca L.): new insights on the organization and the identification of genomic resistance factors. Mol Plant Pathol 10(3):347–360. doi:10.1111/j.1364-3703.2009.00535.x
Mari M, Casalini L, Baraldi E, Bertolini P, Pratella GC (2003) Susceptibility of apricot and peach fruit to Monilinia laxa during phenological stages. Postharvest Biol Technol 30(1):105–109. doi:10.1016/s0925-5214(03)00138-8
Martinez-Garcia PJ, Parfitt DE, Bostock RM, Fresnedo-Ramirez J, Vazquez-Lobo A, Ogundiwin EA, Gradziel TM, Crisosto CH (2013a) Application of genomic and quantitative genetic tools to identify candidate resistance genes for brown rot resistance in peach. PLos ONE 8(11):12. doi:10.1371/journal.pone.0078634
Martinez-Garcia PJ, Parfitt DE, Ogundiwin EA, Fass J, Chan HM, Ahmad R, Lurie S, Dandekar A, Gradziel TM, Crisosto CH (2013b) High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.). Tree Genet Genome 9(1):19–36. doi:10.1007/s11295-012-0522-7
May-De-Mio LL, Tutida I, Motta ACV, Dolinski MA, Serrat BM, Monteguti D (2008) Rate of application of nitrogen and potassium in relation to brown rot and scab in plum ‘Reubennel’ in the region of Araucaia, Parana. Trop Plant Pathol 33(1):35–40
Measham PF, Bound SA, Gracie AJ, Wilson SJ (2009) Incidence and type of cracking in sweet cherry (Prunus avium L.) are affected by genotype and season. Crop Pasture Sci 60(10):1002–1008. doi:10.1071/cp08410
Mengiste T (2012) Plant immunity to necrotrophs. Annu Rev Phytopathol 50(50):267–294. doi:10.1146/annurev-phyto-081211-172955
Messina R, Lain O, Marrazzo MT, Cipriani G, Testolin R (2004) New set of microsatellite loci isolated in apricot. Mol Ecol Notes 4(3):432–434. doi:10.1111/j.1471-8286.2004.00674.x
Mnejja M, Garcia-Mas M, Howad W, Badenes ML, Arus P (2004) Simple-sequence repeat (SSR) markers of Japanese plum (Prunus salicina Lindl.) are highly polymorphic and transferable to peach and almond. Mol Ecol Notes 4(2):163–166. doi:10.1111/j.1471-8286.2004.00603.x
Mnejja M, Garcia-Mas J, Howad W, Arus P (2005) Development and transportability across Prunus species of 42 polymorphic almond microsatellites. Mol Ecol Notes 5(3):531–535. doi:10.1111/j.1471-8286.2005.00977.x
Mnejja M, Garcia-Mas J, Audergon J-M, Arus P (2010) Prunus microsatellite marker transferability across rosaceous crops. Tree Genet Genome 6(5):689–700. doi:10.1007/s11295-010-0284-z
Ogundiwin EA, Peace CP, Nicolet CM, Rashbrook VK, Gradziel TM, Bliss FA, Parfitt D, Crisosto CH (2008) Leucoanthocyanidin dioxygenase gene (PpLDOX): a potential functional marker for cold storage browning in peach. Tree Genet Genome 4(3):543–554. doi:10.1007/s11295-007-0130-0
Pacheco I, Bassi D (2010) Susceptibility to Brown Rot (Monilinia spp.) in Peach (Prunus persica L. Batsch): influence of the maturity date in an F1 progeny. Minerva Biotecnologica Suppl 1(2):57–58
Pascal T, Levigneron A, Kervella J, Nguyenthe C (1994) Evaluation of 2 screening methods for resistance of apricot, plum and peach to Monilinia laxa. Euphytica 77(1–2):19–23
Phillips DJ (1984) Effect of temperature on Monilinia fructicola conidia produced on fresh stone fruit. Plant Dis 68:610–612
Pirona R, Eduardo I, Pacheco I, Linge CDS, Miculan M, Verde I, Tartarini S, Dondini L, Pea G, Bassi D, Rossini L (2013) Fine mapping and identification of a candidate gene for a major locus controlling maturity date in peach. BMC Plant Biology 13. doi:10.1186/1471-2229-13-166
Quilot B, Wu BH, Kervella J, Génard M, Foulongne M, Moreau K (2004) QTL analysis of quality traits in an advanced backcross between Prunus persica cultivars and the wild relative species P. davidiana. Theor Appl Genet 109(4):884–897. doi:10.1007/s00122-004-1703-z
Rant JC, Arraiano LS, Chabannes M, Brown JKM (2013) Quantitative trait loci for partial resistance to Pseudomonas syringae pv. maculicola in Arabidopsis thaliana. Mol Plant Pathol 14(8):828–837. doi:10.1111/mpp.12043
Ritchie DF (2000) Brown rot of stone fruits. Plant Health Instr. doi:10.1094/PHI-I-2000-1025-01
Scorza R, Mehlenbacher SA, Lightner GW (1985) Inbreeding and coancestry of freestone peach cultivars of the Eastern United Sstates and implications for peach germplasm improvement. J Am Soc Hortic Sci 110(4):547–552
Singh R, Tan SG, Panandam JM, Rahman RA, Ooi LCL, Low ETL, Sharma M, Jansen J, Cheah SC (2009) Mapping quantitative trait loci (QTLs) for fatty acid composition in an interspecific cross of oil palm. BMC Plant Biol 9:19. doi:10.1186/1471-2229-9-114
Sosinski B, Gannavarapu M, Hager LD, Beck LE, King GJ, Ryder CD, Rajapakse S, Baird WV, Ballard RE, Abbott AG (2000) Characterization of microsatellite markers in peach Prunus persica (L.) Batsch. Theor Appl Genet 101(3):421–428
Sosinski B, Shulaev V, Dhingra A, Kalyanaraman A, Bumgarner R, Rokhsar D, Verde I, Velasco R, Abbott AG (2009) Rosaceaous genome sequencing: perspectives and progress genetics and genomics of Rosaceae. In: Folta KM, Gardiner SE (eds) Plant genetics and genomics: crops and models, vol 6. Springer, New York, pp 601–615. doi:10.1007/978-0-387-77491-6_28
Stange M, Utz HF, Schrag TA, Melchinger AE, Wurschum T (2013) High-density genotyping: an overkill for QTL mapping? Lessons learned from a case study in maize and simulations. Theor Appl Genet 126(10):2563–2574. doi:10.1007/s00122-013-2155-0
Swaminathan K, Chae WB, Mitros T, Varala K, Xie L, Barling A, Glowacka K, Hall M, Jezowski S, Ming R, Hudson M, Juvik JA, Rokhsar DS, Moose SP (2012) A framework genetic map for Miscanthus sinensis from RNAseq-based markers shows recent tetraploidy. BMC Genomics 13:17. doi:10.1186/1471-2164-13-142
Tamm L, Minder CE, Fluckiger W (1995) Phenological analysis of brown rot blossom blight of sweet cherry caused by Monilinia laxa. Phytopathology 85(4):401–408
Testolin R, Marrazzo T, Cipriani G, Quarta R, Verde I, Dettori MT, Pancaldi M, Sansavini S (2000) Microsatellite DNA in peach (Prunus persica L. Batsch) and its use in fingerprinting and testing the genetic origin of cultivars. Genome 43(3):512–520
Tian SP, Bertolini P (1999) Effect of temperature during conidial formation of Monilinia laxa on conidial size, germination and infection of stored nectarines. J Phytopathol-Phytopathol Z 147(11–12):635–641
Topp B, Sherman W, MCB. R (2008) Low-chill cultivar development. In: D.R. Layne and D.Bassi(eds) The peach: botany, production and uses. CABI, Wallingford, pp 106-138
Van Ooijen JW (2006) Joinmap ® 4, Software for the calculation of genetic linkage maps in experimental populations. Wageningen, Netherlands
Van Ooijen JW (2009) MapQTL ® 6, Software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma B. V, Wageningen, Netherlands
Van Ooijen JW (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res 93(5):343–349. doi:10.1017/s0016672311000279
Vaughan SP, Russell K (2004) Characterization of novel microsatellites and development of multiplex PCR for large-scale population studies in wild cherry, Prunus avium. Mol Ecol Notes 4(3):429–431. doi:10.1111/j.1471-8286.2004.00673.x
Vendramin E, Dettori MT, Giovinazzi J, Micali S, Quarta R, Verde I (2006) A set of EST-SSRs isolated from peach fruit transcriptome and their transportability across Prunus species. Mol Ecol Notes 7(2):307–310. doi:10.1111/j.1471-8286.2006.01590.x
Verde I, Lauria M, Dettori M, Vendramin E, Balconi C, Micali S, Wang Y, Marrazzo MT, Cipriani G, Hartings H, Testolin R, Abbott AG, Motto M, Quarta R (2005) Microsatellite and AFLP markers in the Prunus persica L. (Batsch) x P. ferganensis BC(1)linkage map: saturation and coverage improvement. Theor Appl Genet 111(6):1013–1021. doi:10.1007/s00122-005-0006-3
Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, Gasic K, Micheletti D, Rosyara UR, Cattonaro F, Vendramin E, Main D, Aramini V, Blas AL, Mockler TC, Bryant DW, Wilhelm L, Troggio M, Sosinski B, Aranzana MJ, Arus P, Iezzoni A, Morgante M, Peace C (2012) Development and evaluation of a 9K SNP array for peach by internationally coordinated snp detection and validation in breeding germplasm. PLos ONE 7(4):e35668. doi:10.1371/journal.pone.0035668
Verde I, Abbott AG, Scalabrin S, Jung S, Shu SQ, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F, Zuccolo A, Rossini L, Jenkins J, Vendramin E, Meisel LA, Decroocq V, Sosinski B, Prochnik S, Mitros T, Policriti A, Cipriani G, Dondini L, Ficklin S, Goodstein DM, Xuan PF, Del Fabbro C, Aramini V, Copetti D, Gonzalez S, Horner DS, Falchi R, Lucas S, Mica E, Maldonado J, Lazzari B, Bielenberg D, Pirona R, Miculan M, Barakat A, Testolin R, Stella A, Tartarini S, Tonutti P, Arus P, Orellana A, Wells C, Main D, Vizzotto G, Silva H, Salamini F, Schmutz J, Morgante M, Rokhsar DS, Int Peach Genome I (2013) The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45(5):487–U447. doi:10.1038/ng.2586
Villarino M, Sandin-Espana P, Melgarejo P, De Cal A (2011) High chlorogenic and neochlorogenic acid levels in immature peaches reduce Monilinia laxa infection by interfering with fungal melanin biosynthesis. J Agric Food Chem 59(7):3205–3213. doi:10.1021/jf104251z
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93(1):77–78
Wagner Júnior A, Bassols Raseira MC, Pierobom CR, da Silva JB, Franzon RC (2008) Avaliação de diferentes genótipos de pessegueiro quanto à reação a Monilinia fructicola (Wint.) Honey em frutos. Rev CERES 55(2):83–88
Wagner Junior A, Fabiane KC, Marth Alves De Oliveira JS, Zanela J, Citadin I (2011) Peaches tree genetic divergence for brown rot reaction. Revista Brasileira De Fruticultura 33(1)
Walter M, McLlaren GF, Fraser JA, Frampton CM, Boyd-Wilson KSH, Perry JH (2004) Methods of screening apricot fruit for resistance to brown rot caused by Monilinia spp. Aust Plant Pathol 33(4):541–547. doi:10.1071/ap04062
Wang Y, Georgi LL, Zhebentyayeva TN, Reighard GL, Scorza R, Abbott AG (2002) High-throughput targeted SSR marker development in peach (Prunus persica). Genome 45(2):319–328. doi:10.1139/g01-153
Wilcox WF (1989) Influence of environment and inoculum density on the incidence of brown rot blossom blight of sour cherry. Phytopathology 79(5):530–534
Xu Y, Ma RC, Xie H, Liu JT, Cao MQ (2004) Development of SSR markers for the phylogenetic analysis of almond trees from China and the Mediterranean region. Genome 47(6):1091–1104. doi:10.1139/g04-058
Yamamoto T, Mochida K, Imai T, Shi YZ, Ogiwara I, Hayashi T (2002) Microsatellite markers in peach Prunus persica (L.) Batsch derived from an enriched genomic and cDNA libraries. Mol Ecol Notes 2(3):298–301. doi:10.1046/j.1471-8286.2002.00242.x
Yamamoto T, Yamaguchi M, Hayashi T (2005) An integrated genetic linkage map of peach by SSR, STS, AFLP and RAPD. J Japan Soc Hortic Sc 74(3):204–213
Yang NN, Reighard G, Ritchie D, Okie W, Gasic K (2013) Mapping quantitative trait loci associated with resistance to bacterial spot (Xanthomonas arboricola pv. pruni) in peach. Tree Genet Genome 9(2):573–586. doi:10.1007/s11295-012-0580-x
Yoshimura MA, Luo Y, Ma ZH, Michailides TJ (2004) Sensitivity of Monilinia fructicola from stone fruit to thiophanate-methyl, iprodione, and tebuconazole. Plant Dis 88(4):373–378
Zhang GR, Sebolt AM, Sooriyapathirana SS, Wang DC, Bink M, Olmstead JW, Iezzoni AF (2009) Fruit size QTL analysis of an F-1 population derived from a cross between a domesticated sweet cherry cultivar and a wild forest sweet cherry. Tree Genet Genome 6(1):25–36. doi:10.1007/s11295-009-0225-x
Zhou MX, Johnson P, Zhou GF, Li CD, Lance R (2012) Quantitative trait loci for waterlogging tolerance in a Barley Cross of Franklin x YuYaoXiangTian Erleng and the relationship between waterlogging and salinity tolerance. Crop Sci 52(5):2082–2088. doi:10.2135/cropsci2012.01.0008
Ziosi V, Noferini M, Fiori G, Tadiello A, Trainotti L, Casadoro G, Costa G (2008) A new index based on vis spectroscopy to characterize the progression of ripening in peach fruit. Postharvest Biol Technol 49(3):319–329. doi:10.1016/j.postharvbio.2008.01.017
Acknowledgments
This work was supported by an Italian grant to DB funded by private and public agencies “MAS.PES: apricot and peach breeding by molecular-assisted selection.” Thanks to the Executive Committee of Fruit Breedomics European Project (FP7-265582) and Fondazione Edmund Mach, who allowed the use and provided genotypic data of 26 SNPs included in this study. RP was supported by MIUR FIRB grant RBIP06CTBR. The authors wish to thank M Pesenti, S Femia, A Masini, M Lasala, A Arioli, L Nazzari, and E Quattrini for technical assistance in field and lab operations, as well as Dr. S Biffani for the advice on statistical analyses and Prof. F Faoro for the advice on plant pathology issues.
Author contributions
IP constructed the genetic map, performed and coordinated phenotyping, conducted statistical and QTL analysis and drafted and corrected the manuscript. DB assessed the potential of the plant material, developed the population, helped to draft the manuscript, managed and coordinated the study. IE and RP participated in marker analysis and helped to draft the manuscript. AC helped in phenotyping, QTL analyses, and helped in the revision of the manuscript. LR managed and coordinated the study and helped to draft the manuscript. AV managed and coordinated the study, participated in its design. All authors read and approved the final manuscript.
Data archiving statement
C × EL linkage map and QTL positions will be submitted on Genome Database for Rosaceae (www.rosaceae.org).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E. Dirlewanger
Rights and permissions
About this article

Cite this article
Pacheco, I., Bassi, D., Eduardo, I. et al. QTL mapping for brown rot (Monilinia fructigena) resistance in an intraspecific peach (Prunus persica L. Batsch) F1 progeny. Tree Genetics & Genomes 10, 1223–1242 (2014). https://doi.org/10.1007/s11295-014-0756-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-014-0756-7