
Abstract
The development of single nucleotide polymorphism (SNP) markers in Japanese pear (Pyrus pyrifolia Nakai) offers the opportunity to use DNA markers for marker-assisted selection in breeding programs because of their high abundance, codominant inheritance, and potential for automated high-throughput analysis. We developed a 1,536-SNP bead array without a reference genome sequence from more than 44,000 base changes on the basis of a large-scale expressed sequence tag (EST) analysis combined with 454 genome sequencing data of Japanese pear ‘Housui’. Among the 1,536 SNPs on the array, 756 SNPs were genotyped, and 609 SNP loci were mapped to linkage groups on a genetic linkage map of ‘Housui’, based on progeny of an interspecific cross between European pear (Pyrus communis L.) ‘Bartlett’ and ‘Housui’. The newly constructed genetic linkage map consists of 951 loci, comprising 609 new SNPs, 110 pear genomic simple sequence repeats (SSRs), 25 pear EST–SSRs, 127 apple SSRs, 61 pear SNPs identified by the “potential intron polymorphism” method, and 19 other loci. The map covers 22 linkage groups spanning 1341.9 cM with an average distance of 1.41 cM between markers and is anchored to reference genetic linkage maps of European pears and apples. A total of 514 contigs containing mapped SNP loci showed significant similarity to known proteins by functional annotation analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High-density genetic linkage maps constructed with genome-wide molecular markers are useful for fruit tree breeding programs, as the implementation of marker-assisted selection (MAS) can accelerate the selection process and reduce the progeny size and the cost of raising individuals to maturity in the field (Luby and Shaw 2001). Attempts to use MAS, however, are just beginning and remain limited to the selection of a few simply inherited traits, especially in fruit trees, because marker development for MAS via bi-parental quantitative trait locus (QTL) mapping is hindered by the same complications as phenotyping for traditional breeding. New genomic-based strategies using genome-wide molecular markers such as genome-wide association study (GWAS) and genomic selection (GS) (Meuwissen et al. 2001) have now emerged as powerful tools (Grattapaglia and Resende 2011; Kumar et al. 2012; Kumar et al. 2013).
The family Rosaceae is a medium-sized family of flowering plants, including about 3,000 species in 100 genera. Many economically important products are derived from the Rosaceae, including many edible fruits (apples, apricots, plums, cherries, peaches, pears, raspberries, and strawberries) and ornamental trees and shrubs (Hummer and Janick 2009). Draft genome sequences for apple (Malus × domestica Borkh., Velasco et al. 2010), the woodland strawberry (Fragaria vesca, Shulaev et al. 2011), and peach (Prunus persica (L.) Batsch, http://www.rosaceae.org/) were the first to be reported. Recently, genome sequences have become available of Chinese pear (Pyrus bretschneideri Rehd.) (Wu et al. 2013).
Pears (Pyrus spp.) have been one of the most important fruit trees in Europe, East Asia, and North America for up to 3,000 years and are commercially grown in around 50 temperate-climate countries (Bell et al. 1996). Pear, like the other pipfruit species apple, belongs to the subfamily Spiraeoideae, tribe Pyreae, sharing a basic chromosome number of x = 17 which indicates a polyploid origin. It was shown that a relatively recent (>50 million years ago) genome-wide duplication has resulted in the transition from 9 ancestral chromosomes to 17 chromosomes in the Pyreae and that the ancestral paleohexaploidy of eudicots was supported (Velasco et al. 2010). Pear genotypes are highly heterozygous, because of the self-incompatibility character (Kikuchi 1929).
Owing to these advances in the sequencing of plant genomes, single nucleotide polymorphisms (SNPs) have become a practical choice in genetic analysis and are routinely used as markers in many genetic applications, including the detection of genotype–phenotype associations, the construction of genetic maps, and marker-assisted breeding (Gupta et al. 2001). SNPs are abundant throughout the genome and suited to automated detection (Chen and Sullivan 2003), and represent codominant markers with a simple, well-defined mutation model (Brookes 1999). SNPs have a low mutation rate and fewer detection or evaluation errors than simple sequence repeats (SSRs; Yu et al. 2011) and are often transferable across species within a genus (Grattapaglia et al. 2011). They are far more prevalent than SSRs and therefore may provide a high density of markers near loci of interest. SNPs are now considered useful markers in ecological and evolutionary studies of non-model species (Morin et al. 2004).
Large numbers of SNP markers are currently available for many plant species, including Arabidopsis thaliana (Schmid et al. 2003), maize (Zea mays L.) (Tenaillon et al. 2001), rice (Oryza sativa L.) (Feltus et al. 2004), and soybean (Glycine max Merr.) (Zhu et al. 2003). Among Rosaceae fruit trees, the 8K SNP array developed in apple (Chagné et al. 2012) also includes 1K pear SNPs (Montanari et al. 2013) and there is a 9K SNP array in peach (Verde et al. 2012) and a 6K array in cherry (Peace et al. 2012). However, although pears (Pyrus spp.) are among the most important fruits worldwide, the number of SNP markers developed in pears is still low.
In this study, we developed 1,300 SNPs from expressed sequence tags (ESTs) and 236 SNPs from genome sequences for a GoldenGate assay of Japanese pear ‘Housui’. More than 600 SNPs were used to construct a genetic linkage map of ‘Housui’. Future applications of the new genome-wide SNP markers are discussed.
Materials and methods
Plant materials and extraction of nucleic acids
Japanese pear ‘Housui’ (syn. ‘Hosui’) was used both for RNA sequencing (EST analysis) and genome sequencing. We constructed 11 cDNA libraries, representing leaf bud, leaf, flower bud, flower before opening, and flower at full bloom, as well as fruitlets at three developmental stages: immature fruit, fruit at optimum maturity for eating, and overripe fruit (Nishitani et al. 2009a). Total RNA was extracted by the hot borate method (Wan and Wilkins 1994), and poly(A)+ RNA was purified with a FastTrack 2.0 kit (Invitrogen, USA).
Genomic DNA was isolated from young leaves with a genomic DNA buffer set and Genomic-tip 20/G anion-exchange columns (Qiagen, Germany) as described in Yamamoto et al. (2006).
The progenies of an interspecific cross between European pear (Pyrus communis L.) ‘Bartlett’ and ‘Housui’ (63 F1 plants) were used to construct the genetic linkage map of ‘Housui’ (Terakami et al. 2009). All plant materials were maintained at the NARO Institute of Fruit Tree Science (NIFTS, Ibaraki, Japan).
Pyrosequencing of cDNA and genomic DNA
Approximately 3 μg of adaptor-ligated complementary DNAs (cDNAs) from each of the 11 libraries was sheared via nebulization into small fragments, and then, fragments ranging from 400 to 1,000 bp in length were recovered. cDNAs were sequenced on a Roche/454 Genome Sequencer (GS)-FLX Titanium platform (Roche Diagnostics, Germany; Margulies et al. 2005). Total genomic DNA of ‘Housui’ was sheared by nebulization (600 to 900 bp in length), amplified by emulsion PCR, and pyrosequenced on the Roche/454 GS-FLX Titanium platform as described in Kim et al. (2012).
SNP discovery and bead array construction
The Roche library adaptors were screened and masked with the CROSS_MATCH utility (Ewing and Green 1998; http://www.phrap.org/), using the parameters -minmatch 12 -penalty -2 -minscore 20. All sequences were screened for contaminants and trimmed by the SeqClean script (http://compbio.dfci.harvard.edu/tgi/software/) using the Japanese pear chloroplast genome (Terakami et al. 2012, NCBI accession AP012207), mitochondrial gene sequences of tobacco (Nicotiana tabacum, BA000042), and the Univec database (http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html) with default parameters. Invalid sequences shorter than 40 bp were removed. After screening and trimming, the EST sequences were assembled using the MIRA v. 3.0.5 sequence assembler (Chevreux et al. 2004, http://sourceforge.net/apps/mediawiki/mira-assembler) with the commands -job = denovo,est,normal,454 -fasta -noclipping -notraceinfo -LR:eq = none -AS:mrl = 40 -SK:pr = 98 -AL:mo = 40 -AL:mrs = 95. The genome was assembled by the PCAP.REP assembler (Huang et al. 2006) with default parameters. Contigs of <100 bases in length, singletons, and any unassembled or “debris” sequences were excluded from further analyses. Bi-allelic SNPs identified from contigs using GigaBayes software (Marth et al. 1999) were filtered according to quality value (QV >20), position on the contigs (>100 bp apart), distance from contig ends (>100 bp), read depth of coverage (>6), and allele frequency (0.33–0.67).
To choose SNPs in non-exon regions for an alternative approach, we excluded contigs of genome sequences matching EST contigs. The obtained contig sequences harboring SNPs obtained from both exon and non-exon regions were then submitted to the online Assay Design Tool (ADT; Illumina, USA) for analysis. SNPs with an ADT score below 0.6 were discarded, and 2,786 SNPs on exon and 1,406 SNPs on non-exon regions remained. Finally, a total of 1,536 SNPs with an ADT score of >0.6 were used to design a 1,536-plex custom oligo pool assay (Illumina) (Fig. 1). Of the 1,536 SNPs, 1,300 were positioned in gene exons and the other 236 in non-exon regions. These newly developed SNPs are denoted “JPsnpHou.” All of the pyrosequence data (.sff files) were deposited in DDBJ Sequence Read Archive (DRA001738).

Flowchart for designing the 1,536-plex custom oligo pool assay (OPA). Raw reads obtained by the GS-FLX Titanium platform were treated to mask library adaptors and to exclude low-QV and short reads. The resulting clean reads were assembled with the MIRA or PCAP.REP sequence assembler. Bi-allelic SNPs were identified from assembled contigs using GigaBayes software. Candidate SNPs were filtered according to several criteria
SNP genotyping
SNPs were genotyped using an Illumina GoldenGate Genotyping Assay. The scanned data were analyzed by the Genotyping module (v. 1.9.4) of Illumina GenomeStudio (v. 2011.1) software to generate genotype data for individuals. Clustering of SNPs was adjusted by eye when necessary. Acceptable SNPs had scores of “GenTrain score” ≥ 0.5, “call freq” ≥ 0.85, “P-P-C errors” = 0, and “minor freq” ≥ 0.01.
Genome mapping of SNP markers
Molecular markers used for construction of the genetic linkage map of ‘Housui’—pear SNPs, pear SSRs, apple SSRs, and other markers—are listed in Table 1 as well as the 609 newly designed markers. They include 61 SNP markers developed by the potential intron polymorphism (PIP) method, designed from putative intron information of apple ESTs (Terakami et al. 2013). AFLP markers, which were denoted “EcoR I primer/Mse I primer-fragment size” in the previous linkage map of ‘Housui’ (Terakami et al. 2009), were excluded.
The new ‘Housui’ genetic linkage map was constructed using JoinMap v. 4.0 software (Van Ooijen 2006), with a pseudo-testcross strategy (Grattapaglia and Sederoff 1994). An independence logarithm of odds (LOD) score of 5.0 was used to define linkage groups (LGs). The regression mapping algorithm was used to build the linkage maps, and map distances were calculated according to Kosambi’s mapping function (Kosambi 1944). The linkage map was drawn in MapChart 2.2 software (Voorrips 2002).
The contigs that generated SNPs on the genetic linkage map were functionally annotated by similarity searches against the NCBI non-redundant protein (Nr) database (http://www.ncbi.nlm.nih.gov) using the BLASTX algorithm with an E-value cutoff of 1E–15. A BLASTN analysis against the draft genome of the Chinese pear (P. bretschneideri Rehd., BioProject; PRJNA157875, Pbr_v1.0) was also performed to search for a top hit scaffold associated with the contigs.
Results
De novo assembly of pyrosequenced ESTs and genome DNA
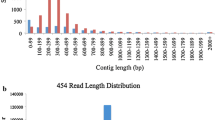
We obtained 497,325 sequences from the 11 cDNA libraries, with an average sequence length of 373.5 bases (range, 40–1,196). They yielded a total sequenced length of 185 Mb. After poly(A/T) and adapter sequences were removed, 484,361 EST sequences (97.4 %) remained for assembly. This generated 46,606 contigs consisting of 359,959 sequences. Contig lengths averaged 573.6 bp (range, 100–2,258). The N50 size was 595 bp. A total of 854 contig sequences (1.8 %) were longer than 1,000 bp. The average depth of contigs was 7.72, and 7,545 contigs (16.2 %) were assembled from more than 10 sequences.
We generated 1,267,079 sequences from the genomic DNA, with an average sequence length of 417.4 bases, yielding a total sequenced length of 529 Mb, which is equivalent to the haploid genome of P. bretschneideri (Wu et al. 2013). Chloroplast and mitochondrial genomes, present at the respective ratios of 2.6 and 1.1 %, were excluded from further analysis. Assembly generated 83,787 contigs consisting of 316,084 sequences. Contig lengths averaged 667.3 bp (range, 100–3,596). The average depth of contigs was 3.77, and 2,753 contigs (3.3 %) were assembled from more than 10 sequences.
SNP discovery and detection
The assembly of cDNA sequences revealed 24,031 base changes in putative heterozygous alleles of ‘Housui’ derived from 8,777 EST contigs. Similarly, the assembly of genome sequences revealed 20,882 base changes in putative heterozygous alleles of ‘Housui’ derived from 7,365 contigs. After filtering, 5,215 candidate SNPs remained.
Optimization resulted in 2,786 SNPs from exon regions (ESTs) and 1,406 SNPs from non-exon regions (genome sequences). From these, 1,536 SNPs were selected (ADT score >0.60; mean, 0.86). Each SNP was chosen from a unique contig. Of the 1,536 SNPs, 1,300 were developed from exon regions and 236 from non-exon regions.
SNP genotyping and mapping
A total of 756 SNPs from the 1,536-SNP GoldenGate bead array were successfully genotyped and exhibited a heterozygous genotype. Among them, 617 were heterozygous for ‘Housui’ and homozygous for ‘Bartlett’, and showed clear segregation within the mapping population. The remaining SNPs were unmapped or ungenotyped. Linkage analysis allowed 609 of these markers to be located in ‘Housui’ LGs. The remaining eight markers were not mapped to LGs. Detailed information on the mapped SNPs is given in supplementary materials (Table S1).
The new genetic linkage map of ‘Housui’ consists of 951 loci, comprising 609 new SNPs (denoted as JPsnpHou), 110 pear genomic SSRs, 25 pear EST–SSRs, 127 apple SSRs, 61 pear SNPs determined by the PIP method, and 19 other loci (Fig. 2). The map covers 22 LGs spanning 1,341.9 cM with an average distance of 1.41 cM between markers (Table 2). Twenty LGs are anchored to the reference genetic linkage maps of European pear (Yamamoto et al. 2007) and apple (Celton et al. 2009a; Liebhard et al. 2003; Silfverberg-Dilworth et al. 2006). Subgroups of three LGs (Ho2, Ho5, and Ho12), with an independence LOD score of 3.0, are not grouped. Two LGs (uk-1 and uk-2) are not anchored to any LGs of reference maps. The mapped SNP markers seem to be distributed among all LGs without bias. Segregation of 127 loci was distorted (P < 0.05), and many molecular marker loci in LGs Ho2-1 and Ho2-2 were distorted.



Genetic linkage map of Japanese pear Housui. The number to the left of each marker indicates genetic distance (cM). Markers designated in green are developed from pear and markers in red from apple. New markers developed in this study are denoted by JPsnpHou. Distorted segregation is indicated by a significant P value of the χ 2 test: *P = 0.05, **P = 0.01, ***P = 0.005
Discussion
High-throughput systems for screening genetic markers are essential for genetic studies and plant breeding. In the Rosaceae, the International RosBREED SNP Consortium developed an 8K genome-wide SNP array for apple (Chagné et al. 2012), selecting 27 apple cultivars to represent worldwide breeding germplasm and re-sequencing them at low coverage. Alignment of these sequences with the whole genome sequence of ‘Golden Delicious’ (Velasco et al. 2010) enabled the consortium to establish 7,867 apple SNPs (Chagné et al. 2012). Khan et al. (2012) developed a high-throughput 1,536-EST-derived SNP GoldenGate genotyping platform in apple, containing 1,411 genic SNPs and 125 genomic SNPs. The International Peach SNP Consortium re-sequenced the whole genome of 56 peach breeding accessions through the use of next-generation sequencing platforms (Verde et al. 2012) and developed a 9K SNP array (Verde et al. 2012). Martínez-García et al. (2013) evaluated a set of 1,536 SNPs through a GoldenGate genotyping assay of peach (P. persica (L.) Batsch), which was developed from the whole genome sequences of three cultivars. The RosBREED Consortium also developed a 6K SNP array for diploid sweet cherry (Prunus avium) and allotetraploid sour cherry (Prunus cerasus) (Peace et al. 2012). Our large-scale transcriptome-based analysis of Japanese pear in conjunction with genome sequencing data allowed us to develop a 1,536-SNP array without a reference genome sequence. It will be interesting to integrate our transcriptome-based analysis and Chinese pear genome information. Montanari et al. (2013) reported that 1,096 SNPs were developed from three European pear cultivars by using new-generation sequencing technology and that SNPs obtained showed high transferability across several Pyrus spp. Our 609 new SNP markers will complement the 829 SNP markers reported in Montanari et al. (2013) to segregate in interspecific Pyrus progenies, which enable the discovery of associations between marker loci and traits, the identification of the genetic architecture of quantitative traits, investigation of genetic variation, and GS in pear.
Of the 1,536 SNP loci, 609 (39.6 %) were successfully validated for genome mapping, resulting in the development of a high-density genetic linkage map comprising 951 loci. The remaining SNPs were ungenotyped or unmapped, resulting from several reasons, i.e., the unexpected homozygous allele combination for ‘Housui’ (27.6 %), the heterozygous genotypes for both parents of ‘Housui’ and ‘Bartlett’ (9.0 %), unassigned SNPs to 17 LGs (0.5 %), ambiguous clustering (4.5 %), and the low signal for scoring (18.7 %). A rather low success ratio of SNP mapping (39.6 %) in our study may be improved by the use of transcriptome and genome sequencing for plural cultivars. Furthermore, only 8 out of 256 SNPs (3.1 %) developed from non-exon regions that could be positioned on the ‘Housui’ map, compared with 601 out of 1,300 SNPs (46.2 %) that showed successful genome mapping from exon regions. This may be consistent with the report that the randomly distributed SNPs tended to show a lower success rate than SNPs located within coding regions in the apple 8K SNP array (Chagné et al. 2012). Mapping of new SNP markers enables the newly constructed map to increase about threefold in marker loci, decrease to two fifths in average distance between markers, and expand 168 cM in map distance, compared with the previous map (Terakami et al. 2009). In our previous report, three particular genomic regions (LGs 4, 5, and 12) were identified showing short length of map distance, which may be due to homozygous regions (Terakami et al. 2009). The new map could expand the three LGs, from 9.7 to 18.5 cM in LG 4, from 23.2 to 48.9 cM in LG 5, and from 0 to 27.5 cM in LG 12, even if LGs 5 and 12 were divided into two parts. The GoldenGate assay has also been used to evaluate SNPs for the creation of high-density genetic linkage maps in other crops: Khan et al. (2012) mapped 569 new SNPs of 1,536 GoldenGate genotyping platform along with 447 other markers, constructing of new genetic maps for ‘Co-op 16’ and ‘Co-op 17’ of apple. Khan et al. (2012) also reported that markers common across five apple genetic maps resulted in successful positioning of 2,875 markers, consisting of 2,033 SNPs and 843 SSRs, and that the total length of the consensus map was 1,991 cM. Martínez-García et al. (2013) identified 738 of 1,037 SNP markers on the Pop-DF map of peach. Shirasawa et al. (2010) mapped 1,137 markers, including 793 genotyped SNPs, in two mapping populations of tomato. Hyten et al. (2008) designed a custom 384-SNP assay through the re-sequencing of five soybean accessions, evaluated it with three mapping populations of recombinant inbred lines, and genotyped 89 % of the SNP loci in the complex soybean genome. Jones et al. (2009) used a 768-marker multiplex assay on the GoldenGate platform to create a high-resolution genetic map in maize.
SNPs have several advantages over other molecular markers, including their high abundance in the genome, codominant mode of inheritance, and high integrity. In the previous study, we applied apple PIP marker information (Wu et al. 2007) to develop SNPs in Japanese pear (Terakami et al. 2013), since apple and pear, which belong to the same tribe, Pyreae, showed genome-wide synteny (Yamamoto et al. 2001; Silfverberg-Dilworth et al. 2006; Celton et al. 2009b). The use of PIP markers designed from apple ESTs enabled the mapping of 55 markers on the genetic linkage map of ‘Bartlett’ and 61 in ‘Housui’. However, in the present study, we established a more robust approach for developing the larger numbers of genome-wide SNP markers that will be needed for introducing GWAS and GS into Japanese pear breeding.
Kumar et al. (2012) evaluated the accuracy of GS in a population of seven full-sib families comprising 1,120 seedlings genotyped for 2,500 SNPs using the 8K apple SNP array and phenotyped for six fruit quality-related traits (fruit firmness, soluble solids, russet, weighted cortex intensity, astringency, titratable acidity). They observed accuracies ranging from 0.67 (astringency) to 0.89 (soluble solids) for BLUP-based selection, demonstrating that GS is a credible alternative to conventional selection for fruit quality traits. It was suggested that a higher SNP density and a larger training population might capture more genetic variation with higher accuracy (Kumar et al. 2013). Kumar et al. (2013) tried GWAS using the same genotyped 1,200 apple seedlings, demonstrating that the genomic regions could be identified with significant effects for several fruit quality traits and pointing out the value of GWA-significant SNP–trait associations in a breeding population for MAS. Iwata et al. (2013) examined the potential of GWAS and GS using 76 Japanese pear cultivars genotyped with 162 DNA markers including 155 SSRs and phenotyped for nine agronomic traits, using multilocus Bayesian models for analysis in Japanese pear. Correlations between phenotypic values and predicted genotypic values were significantly detected for harvest time, resistance to black spot, and the number of spurs with the accuracies of 0.75, 0.38, and 0.61, respectively. Two associations to known loci for resistance to black spot and harvest time were correctly identified. It was noted that neither the number of markers nor genotypes used were sufficient to conduct full-scale GWAS and to train a prediction model for GS. The more than 600 SNP markers newly obtained are a valuable resource which will enable the implementation of MAS and will contribute to complete the further application of GWAS and GS in Japanese pear breeding programs.
References
Bell RL, Quamme HA, Layne REC, Skirvin RM (1996) Pears. In: Janick J, Moore JN (eds) Fruit breeding, volume I: tree and tropical fruits. Wiley, New York, pp 441–514
Brookes AJ (1999) The essence of SNPs. Gene 234:177–186
Celton JM, Tustin DS, Chagné D, Gardiner SE (2009a) Construction of a dense genetic linkage map for apple rootstocks using SSRs developed from Malus ESTs and Pyrus genomic sequences. Tree Genet Genomes 5:93–107
Celton JM, Chagné D, Tustin SD, Terakami S, Nishitani C, Yamamoto T, Gardiner SE (2009b) Update on comparative genome mapping between Malus and Pyrus. BMC Research Notes 2:182
Chagné D, Crowhurst RN, Troggio M, Davey MW, Gilmore B, Lawley C, Vanderzande S, Hellens RP, Kumar S, Cestaro A et al (2012) Genome-wide SNP detection, validation, and development of an 8 K SNP array for apple. PLoS One 7:e31745
Chen X, Sullivan PF (2003) Single nucleotide polymorphism genotyping: biochemistry, protocol, cost and throughput. Pharmacogenomics J 3:77–96
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WEG, Wetter T, Suhai S (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14:1147–1159
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Feltus FA, Wan J, Schulze SR, Estill JC, Jiang N, Paterson AH (2004) An SNP resource for rice genetics and breeding based on subspecies Indica and Japonica genome alignments. Genome Res 14:1812–1819
Fernandez-Fernandez F, Harvey NG, James CM (2006) Isolation and characterization of polymorphic microsatellite markers from European pear (Pyrus communis L.). Mol Ecol Notes 6:1039–1041
Grattapaglia D, Resende MDV (2011) Genomic selection in forest tree breeding. Tree Genet Genomes 7:241–255
Grattapaglia D, Sederoff R (1994) Genetic-linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Grattapaglia D, Silva OB, Kirst M, de Lima BM, Faria DA, Pappas GJ (2011) High-throughput SNP genotyping in the highly heterozygous genome of Eucalyptus: assay success, polymorphism and transferability across species. BMC Plant Biol 11:65
Guilford P, Prakash S, Zhu JM, Rikkerink E, Gardiner S, Bassett H, Forster R (1997) Microsatellites in Malus × domestica (apple): abundance, polymorphism and cultivar identification. Theor Appl Genet 94:249–254
Gupta PK, Roy JK, Prasad M (2001) Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants. Curr Sci India 80:524–535
Huang XQ, Yang SP, Chinwalla AT, Hillier LW, Minx P, Mardis ER, Wilson RK (2006) Application of a superword array in genome assembly. Nucleic Acids Res 34:201–205
Hummer KE, Janick J (2009) Rosaceae: taxonomy, economic importance, genomics. In: Folta KM, Gardiner SE (eds) Genetics and genomics of Rosaceae. Springer, New York, pp 1–17
Hyten DL, Song Q, Choi IY, Yoon MS, Specht JE, Matukumalli LK, Nelson RL, Shoemaker RC, Young ND, Cregan PB (2008) High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor Appl Genet 116:945–952
Inoue E, Matsuki Y, Anzai H, Evans K (2007) Isolation and characterization of microsatellite markers in Japanese pear (Pyrus pyrifolia Nakai). Mol Ecol Notes 7:445–447
Iwata H, Hayashi T, Terakami S, Takada N, Sawamura Y, Yamamoto T (2013) Potential assessment of genome-wide association study and genomic selection in Japanese pear Pyrus pyrifolia. Breed Sci 63:125–140
Jones E, Chu WC, Ayele M, Ho J, Bruggeman E, Yourstone K, Rafalski A, Smith O, McMullen M, Bezawada C et al (2009) Development of single nucleotide polymorphism (SNP) markers for use in commercial maize (Zea mays L.) germplasm. Mol Breed 24:165–176
Khan MA, Han Y, Zhao YF, Troggio M, Schuyler S, Korban SS (2012) A multi-population consensus genetic map reveals inconsistent marker order among maps likely attributed to structural variations in the apple genome. PLoS One 7:e47864
Kikuchi A (1929) Investigation in 1927 and 1928. 1. Paterclinical incompatibility in Japanese pear. J Okitsu Hort Soc 24:1–6 (in Japanese)
Kim H, Terakami S, Nishitani C, Kurita K, Kanamori H, Katayose Y, Sawamura Y, Saito T, Yamamoto T (2012) Development of cultivar-specific DNA markers based on retrotransposon-based insertional polymorphism in Japanese pear. Breed Sci 62:53–62
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugenic 12:172–175
Kumar S, Chagné D, Bink MCAM, Volz RK, Whitworth C, Carlisle C (2012) Genomic selection for fruit quality traits in apple (Malus × domestica Borkh.). PLoS One 7:e36674
Kumar S, Garrick DJ, Bink MCAM, Whitworth C, Chagné D, Volz RK (2013) Novel genomic approaches unravel genetic architecture of complex traits in apple. BMC Genomics 14:393
Liebhard R, Gianfranceschi L, Koller B, Ryder CD, Tarchini R, Van de Weg E, Gessler C (2002) Development and characterisation of 140 new microsatellites in apple (Malus × domestica Borkh.). Mol Breed 10:217–241
Liebhard R, Koller B, Gianfranceschi L, Gessler C (2003) Creating a saturated reference map for the apple (Malus × domestica Borkh.) genome. Theor Appl Genet 106:1497–1508
Luby JJ, Shaw DV (2001) Does marker-assisted selection make dollars and sense in a fruit breeding program? HortSci 36:872–879
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Marth GT, Korf I, Yandell MD, Yeh RT, Gu ZJ, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR (1999) A general approach to single-nucleotide polymorphism discovery. Nat Genet 23:452–456
Martínez-García PJ, Parfitt DE, Ogundiwin EA, Fass J, Chan HM, Ahmad R, Lurie S, Dandekar A, Gradziel TM, Crisosto CH (2013) High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.). Tree Genet Genomes 9:19–36
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Montanari S, Saeed M, Knäbel M, Kim YK, Troggio M, Malnoy M, Velasco R, Fontana P, Won KH, Durel CE, Perchepied L, Schaffer R, Wiedow C, Bus V, Brewer L, Gardiner SE, Crowhurst RN, Chagné D (2013) Identification of Pyrus single nucleotide polymorphisms (SNPs) and evaluation for genetic mapping in European pear and interspecific Pyrus hybrids. PLoS One 7:e36674
Morin PA, Luikart G, Wayne RK, Grp SW (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216
Moriya S, Iwanami H, Kotoda N, Haji T, Okada K, Terakami S, Mimida N, Yamamoto T, Abe K (2012) Aligned genetic linkage maps of apple rootstock cultivar ‘JM7’ and Malus sieboldii ‘Sanashi 63’ constructed with novel EST–SSRs. Tree Genet Genomes 8:709–723
Nishitani C, Shimizu T, Fujii H, Terakami S, Yamamoto T (2009a) Analysis of expressed sequence tags from Japanese pear ‘Housui’. Acta Hort 814:645–649
Nishitani C, Terakami S, Sawamura Y, Takada N, Yamamoto T (2009b) Development of novel EST–SSR markers derived from Japanese pear (Pyrus pyrifolia). Breed Sci 59:391–400
Peace C, Bassil N, Main D, Ficklin S, Rosyara UR, Stegmeir T, Sebolt A, Gilmore B, Lawley C, Mockler TC, Bryant DW, Wilhelm L, Iezzoni A (2012) Development and evaluation of a genome-wide 6 K SNP array for diploid sweet cherry and tetraploid sour cherry. PLoS One 7:e48305
Schmid KJ, Sorensen TR, Stracke R, Torjek O, Altmann T, Mitchell-Olds T, Weisshaar B (2003) Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res 13:1250–1257
Shirasawa K, Isobe S, Hirakawa H, Asamizu E, Fukuoka H, Just D, Rothan C, Sasamoto S, Fujishiro T, Kishida Y, Kohara M, Tsuruoka H, Wada T, Nakamura Y, Sato S, Tabata S (2010) SNP discovery and linkage map construction in cultivated tomato. DNA Res 17:381–391
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP et al (2011) The genome of woodland strawberry (Fragaria vesca). Nat Genet 43:109–116
Silfverberg-Dilworth E, Matasci CL, Van de Weg WE, Van Kaauwen MPW, Walser M, Kodde LP, Soglio V, Gianfranceschi L, Durel CE, Costa F et al (2006) Microsatellite markers spanning the apple (Malus × domestica Borkh.) genome. Tree Genet Genomes 2:202–224
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS (2001) Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc Natl Acad Sci U S A 98:9161–9166
Terakami S, Kimura T, Nishitani C, Sawamura Y, Saito T, Hirabayashi T, Yamamoto T (2009) Genetic linkage map of the Japanese pear ‘Housui’ identifying three homozygous genomic regions. J Jpn Soc Hort Sci 78:417–424
Terakami S, Matsumura Y, Kurita K, Kanamori H, Katayose Y, Yamamoto T, Katayama H (2012) Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): genome structure and comparative analysis. Tree Genet Genomes 8:841–854
Terakami S, Nishitani C, Yamamoto T (2013) Development of SNP markers for marker-assisted selection in pear. Acta Hort 976:463–469
Van Ooijen JW (2006) JoinMap 4, software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D et al (2010) The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet 42:833–839
Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, Gasic K, Micheletti D, Rosyara UR, Cattonaro F, Vendramin E et al (2012) Development and evaluation of a 9 K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One 7:e35668
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wan CY, Wilkins TA (1994) A modified hot borate method significantly enhances the yield of high-quality RNA from cotton (Gossypium hirsutum L.). Anal Biochem 223:7–12
Wu WR, Yang L, Jin GL, Zhao XQ, Zheng Y, Xu ZH (2007) PIP: a database of potential intron polymorphism markers. Bioinformatics 23:2174–2177
Wu J, Wang ZW, Shi ZB, Zhang S, Ming R, Zhu SL, Khan MA, Tao ST, Korban SS, Wang H et al (2013) The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res 23:396–408
Yamamoto T, Kimura T, Sawamura Y, Kotobuki K, Ban Y, Hayashi T, Matsuta N (2001) SSRs isolated from apple can identify polymorphism and genetic diversity in pear. Theor Appl Genet 102:865–870
Yamamoto T, Kimura T, Shoda M, Imai T, Saito T, Sawamura Y, Kotobuki K, Hayashi T, Matsuta N (2002) Genetic linkage maps constructed by using an interspecific cross between Japanese and European pears. Theor Appl Genet 106:9–18
Yamamoto T, Kimura T, Hayashi T, Ban Y (2006) DNA profiling of fresh and processed fruits in pear. Breed Sci 56:165–171
Yamamoto T, Kimura T, Terakami S, Nishitani C, Sawamura Y, Saito T, Kotobuki K, Hayashi T (2007) Integrated reference genetic linkage maps of pear based on SSR and AFLP markers. Breed Sci 57:321–329
Yamamoto T, Terakami S, Moriya S, Hosaka F, Kurita K, Kanamori H, Katayose Y, Saito T, Nishitani C (2013) DNA markers developed from genome sequencing analysis in Japanese pear (Pyrus pyrifolia). Acta Hort 976:477–483
Yu HH, Xie WB, Wang J, Xing YZ, Xu CG, Li XH, Xiao JH, Zhang QF (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 6:e17595
Zhu YL, Song QJ, Hyten DL, Van Tassell CP, Matukumalli LK, Grimm DR, Hyatt SM, Fickus EW, Young ND, Cregan PB (2003) Single-nucleotide polymorphisms in soybean. Genetics 163:1123–1134
Acknowledgments
We thank Ms. F. Hosaka, N. Shigeta, and N. Yagihashi for their technical help. This work was partially supported by a grant from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation, SGE-1003).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E. Dirlewanger
Data submission
All of the pyrosequence data (.sff files) were deposited in DDBJ Sequence Read Archive (DRA001738).
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(XLSX 216 kb)
Rights and permissions
About this article
Cite this article
Terakami, S., Nishitani, C., Kunihisa, M. et al. Transcriptome-based single nucleotide polymorphism markers for genome mapping in Japanese pear (Pyrus pyrifolia Nakai). Tree Genetics & Genomes 10, 853–863 (2014). https://doi.org/10.1007/s11295-014-0726-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-014-0726-0