
Abstract
A Populus deltoides × Populus trichocarpa F1 pedigree was analyzed for quantitative trait loci (QTLs) affecting ectomycorrhizal development and for microarray characterization of gene networks involved in this symbiosis. A 300 genotype progeny set was evaluated for its ability to form ectomycorrhiza with the basidiomycete Laccaria bicolor. The percentage of mycorrhizal root tips was determined on the root systems of all 300 progeny and their two parents. QTL analysis identified four significant QTLs, one on the P. deltoides and three on the P. trichocarpa genetic maps. These QTLs were aligned to the P. trichocarpa genome and each contained several megabases and encompass numerous genes. NimbleGen whole-genome microarray, using cDNA from RNA extracts of ectomycorrhizal root tips from the parental genotypes P. trichocarpa and P. deltoides, was used to narrow the candidate gene list. Among the 1,543 differentially expressed genes (p value ≤ 0.05; ≥5.0-fold change in transcript level) having different transcript levels in mycorrhiza of the two parents, 41 transcripts were located in the QTL intervals: 20 in Myc_d1, 14 in Myc_t1, and seven in Myc_t2, while no significant differences among transcripts were found in Myc_t3. Among these 41 transcripts, 25 were overrepresented in P. deltoides relative to P. trichocarpa; 16 were overrepresented in P. trichocarpa. The transcript showing the highest overrepresentation in P. trichocarpa mycorrhiza libraries compared to P. deltoides mycorrhiza codes for an ethylene-sensitive EREBP-4 protein which may repress defense mechanisms in P. trichocarpa while the highest overrepresented transcripts in P. deltoides code for proteins/genes typically associated with pathogen resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The majority of terrestrial plants live in association with symbiotic fungi which facilitate their access to soil nutrients. The ectomycorrhizal (ECM) symbiosis is the most common association in forest under boreal and temperate climates. ECM symbioses represent complex biological systems involving numerous multifaceted interactions between the two partners (Martin et al. 2003; Martin and Nehls 2009; Nehls et al. 2001; Peterson and Bonfante 1994). Ectomycorrhizal establishment also depends on edaphic and climatic conditions (Smith and Reads 1997), as well as genetic traits of the partners (Rosado et al. 1994; Tagu et al. 2001).
Poplars are pioneer trees living in riparian ecosystems. They are associated with a great variety of arbuscular mycorrhizal fungi and ectomycorrhizal fungi without which they could not colonized these disturbed habitats. Selected poplar exhibiting a large susceptibility to ectomycorrhizal colonization could have an advantage when planting on to disturbed sites. But the absence of ectomycorrhizal inoculum in these types of sites could be a disadvantage, requiring artificial inoculation. In richer soils, like previously cleared agricultural sites, the selection of clones less dependent on ectomycorrhizal colonization could be another strategy.
The development and functioning of ectomycorrhizas involve the expression of numerous genes encoding for developmental proteins and/or regulatory enzymes both in the fungi and their partners (Martin et al. 2007). The genetic predisposition and molecular adaptation needed for mycorrhizal establishment most likely involve the combined action of numerous gene networks (Martin and Nehls 2009). One quantitative trait loci (QTL) related to ectomycorrhizal formation was located on Populus trichocarpa linkage group IIIa also containing a QTL involved in the pathogenic interaction with the fungus Melampsora larici-populina, the causal agent of leaf rust (Tagu et al. 2005). The recent release of genomic resources, such as the Laccaria bicolor genome (Martin et al. 2008) and the P. trichocarpa genome (Tuskan et al. 2006), will facilitate the identification of genetic factors involved in these complex interactions.
Here, we (1) confirmed and extended the previous QTL analysis (Tagu et al. 2005) by positioning QTLs involved in root colonization by L. bicolor on two improved Populus genetic maps, using an extended progeny set (300 F1 individuals), (2) anchored the QTL markers on the P. trichocarpa genomic sequence, and (3) created a candidate gene list from these QTL intervals. Using a whole-genome oligoarray of P. trichocarpa (Tuskan et al. 2006), we also compared the differential gene expression between P. trichocarpa and Populus deltoides ECMs. Finally, we determined whether genes showing a differential expression between the parents exhibiting a low ectomycorrhizal colonization (P. deltoides) and the parent exhibiting a high ectomycorrhizal colonization (P. trichocarpa) were located in QTL intervals related to ectomycorrhizal colonization.
Material and methods
Biological materials
Plant material consisted of 300 clonally replicated F1 progeny from an interspecific P. deltoides (female parent from Illinois, no. 73028-62) by P. trichocarpa (male parent from Washington, no. 101-74) control-cross family (Family 54B, Pop2; Lefèvre et al. 1998; Tagu et al. 2001; Tagu et al. 2005). Ectomycorrhizal formation was evaluated by inoculating the two parents and the 300 progeny with L. bicolor S238N (Di Battista et al. 1996; Tagu et al. 2001). The 300 F1 genotypes were chosen at random among the 336 genotypes used for the construction of the genetic maps (Jorge et al. 2005). Moreover, these 300 genotypes included the 90 genotypes used for the construction of the initial Pop2 genetic map (Faivre-Rampant et al. 1999). The L. bicolor S238N fungal strain, issued from the INRA-Nancy collection of ectomycorrhizal fungi, was maintained on Pachlewski’s medium (Pachlewski and Pchlevska 1974). This model fungal strain was chosen for its ability to form ectomycorrhizas with Populus and for the availability of its genome and transcriptome (Martin et al. 2008; Peter et al. 2003; Tagu et al. 2001). Mycelium was produced on a peat–vermiculite nutrient mixture and grown in glass jars for 2 months in the dark at 25°C and kept at 4°C before use (Duponnois and Garbaye 1991).
Inoculation
Internode cuttings of each progeny and the two parents were rooted and individually inoculated at the same time in 1–l pots containing a mixture of fungal inoculum (1:9; v/v) and calcinated attapulgite (Oil Dri US Special, Damolin, Denmark; http://www.damolin.dk). Inoculated cuttings were grown for three-and-a-half months in a greenhouse at 15–28°C, photoperiod 12 h at INRA-Nancy (48°41′37″ N, 6°11′05″ E) during spring. Cuttings were watered twice daily and received a weekly nutrient application as described in Frey-Klett et al. (1997). In order to control environmental heterogeneity of the greenhouse, eight clonal replicates for each genotype were randomly distributed in eight blocks. Each block contained one rooted cutting of each of the 300 progeny and the two parents.
Measurements
The percentage of mycorrhizal colonization, as described by Tagu et al. (2001; 2005) was determined three-and-a-half months after inoculation by eight observers. All the observers observed randomly the plants within eight blocks and block after block. Each root system was rinsed with tap water, cut in 1-cm pieces and analyzed under a dissecting microscope. For each root system, 100 apices were randomly examined and assessed as mycorrhizal or non-mycorrhizal. Approximately 25,000 colonized root tips were examined.
Statistical analysis
The percentage of mycorrhizal colonization was transformed using an arcsine √X/100 function prior to variance analysis (ANOVA). Clonal variation was tested by a two-factor ANOVA according to \( yij = \mu + gi + bj + eij \), where yij is the observation value of the ith clone in the jth block, μ is the overall mean, gi is the genetic effect of the ith clone, bj the block effect of the jth block, and eij is the error term (Table 1). The following mixed linear model was applied on an individual cutting basis to detect significant differences in mycorrhizal colonization among the clones:
where, μ is the overall mean, B is the block effect (fixed), G is the genotype effect (random), O is the observer effect (fixed), and ε is the random pooled residual error. Restricted maximum likelihood estimates of genetic, block, and residual variance components (σ 2 G , σ 2 B , and σ 2ε ) were computed, and for each trait, individual broad-sense heritability (h 2) was estimated as:
where, n is the average number of replicates per genotype. Standard deviations (SD) of h 2 were derived from classic estimation of SD for a ratio x/y where x = σ2 G and \( y = \sigma_G^2 + \sigma_{\varepsilon }^2/n \). All analyses were performed with the statistical programs JMP 5.0 (SAS Institute Inc., Cary, NC, USA) and R version 2.9.1 (R. Development Core Team, 2003, www.R-project.org). The genetic coefficient of variation (CVG) was used to compare the relative amounts of genetic variation of traits with different means (Cornelius 1994):
Molecular data and QTL detection
Populus genotyping was conducted using RFLP, STS, RAPD, and microsatellite (SSR) markers in a subset of 90 genotypes. SSR and AFLP genotyping was then extended to 253 additional genotypes (Jorge et al. unpublished). RFLP and STS genotyping was conducted as described by Bradshaw and Stettler (1993). RFLP markers were coded by the letter “P” followed by a 3- to 4-digit number (e.g., P1273). RAPD genotyping was performed according to Villar et al. (1996). RAPD markers were coded by the Operon primer name followed by the molecular weight of the polymorphic band (e.g., M02-1150). AFLP genotyping was performed as described in Jorge et al. (2005). AFLP markers were named after the code of the EcoRI/MseI combination followed by the band ranking on the gel (e.g., E5M5-7). The SSR primers came from two different sources: (1) the International Populus Genome Consortium, SSR named “PMGC” (http://www.ornl.gov/sci/ipgc/ssr_resource.htm), “ORPM” (Tuskan et al. 2004), and “WPMS” (Van der Schoot et al. 2000) and (2) SSR named “ai,” “bi,” and “bu” (Jorge et al. unpublished) developed from public EST databases. Different labeling techniques were successively used for SSR genotyping: primers were labeled with γ-[33P] ATP, with forward fluorochrome-labeled primer or the M13 tailing strategy (Schuelke 2000) as described by Jorge et al. (2005). PCR reactions were performed in a total volume of 10 μl containing 10 mM Tris–HCl, pH 8.3; 50 mM KCl; 1.5 mM MgCl2; 200 μM each of dATP, dCTP, dGTP, and dTTP; 0.2 unit Taq Polymerase (Invitrogen); 5 pmoles reverse primer; 0.5 pmoles M13-tailed forward primer; 5 pmoles M13-labeled primer; and 40 ng DNA. PCR reactions were conducted with 42 cycles of a 30-s denaturation at 94°C, 30 s annealing at 55°C or 52°C, and 30 s extension at 72°C. Each forward primer was 5′-tailed with the M13 forward consensus sequence. The M13-tailed forward primers were then used in combination with a standard M13 primer labeled with fluorescent dye (6FAM, HEX, or NED) at its 5′-end. The amplicons for each SSR marker were separately produced, diluted, and pooled post-PCR by three-color multiplexes (6FAM, HEX, and NED) for polymorphism screening. SSR polymorphisms were visualized using an ABI PRISM 3,100 genetic analyzer (PE Applied Biosystems, Foster City, CA). SSR allele lengths were recorded by GeneScan and Genotyper or GeneMapper softwares.
Markers significantly deviating from Mendelian segregation ratios [1:1] were eliminated from the linkage analysis. Linkage maps were constructed using Mapmaker version 3.0b (Lander and Botstein 1989) and were based on the pseudo-testcross strategy which resulted in the construction of two parental maps (Grattapaglia and Sederoff 1994). Steps for map construction were the same as in Jorge et al. (2005) except that the “Error detection” option was enabled.
QTL analysis
QTLs were determined with means corrected for observer effects as input data and using MultiQTL 2.4 (http://www.multiqtl.com/; Haïfa, Israel). Observer effects for data used in the QTL analysis were corrected using a logistic regression. The option “marker restoration” was used to reduce the effect of missing data. The Kosambi mapping function was chosen for recalculation of maps on genotypic data. Single trait analysis was performed using a combination of interval mapping and multiple interval mapping. That is, the entire genome was first scanned using the one QTL model then a two-linked QTL model was used. Permutation tests (1,000 runs), comparing hypotheses H1—there is one QTL on the chromosome vs. H0—no QTL on the chromosome, were run to obtain chromosome-wise statistical significance. For chromosomes for which a single QTL was detected, permutation tests (1,000 runs) were run to compare the hypotheses H2—two-linked QTLs in the chromosome vs. H1. Subsequently, when p (H2vsH1) ≤ 0.05, permutations were run to compare H2 vs. H0. A two-linked QTL model was only accepted, if the two intervals were not adjacent to each other. To reduce calculation time, full permutations for the two-linked QTL models were only conducted with 1,000 runs when the p value was ≤0.1 after 100 runs. In a last step, multiple interval mapping was performed including all the significant intervals mapping QTLs (single and two-linked QTLs). A QTL was declared significant when p ≤ 0.05. For the remaining significant QTLs, permutations were run per chromosome, using p value thresholds of 0.05 per chromosome. Bootstrap analysis was performed to estimate the 95% confidence interval. QTL analysis was performed with the genetic maps of P. deltoides and P. trichocarpa (Jorge et al. 2005).
RNA extraction and cDNA synthesis
Ectomycorrhizal root tips from P. trichocarpa and P. deltoides inoculated with L. bicolor S238N were harvested and instantly frozen in liquid nitrogen. Total RNA was extracted from three biological replicates (3 × 2 ECMs) of each genotype using the RNeasy micro kit (ref.74004, QIAGEN Courtaboeuf, France) following manufacturer’s instructions. An in-column digestion step with DNAse I (ref.79254, QIAGEN Courtaboeuf, France) was included in the RNA purification. RNA quality was verified using Experion High sens Capillary gels (BIO-RAD, France). The cDNAs for NimbleGen microarrays were synthesized using the Super Smart cDNA Synthesis kit (ref.635000, Clontech-Takara Bio Saint-Germain-en-Laye, France).
NimbleGen microarrays
The Populus whole-genome expression array version 2.0 (DiFazio et al. unpublished data) manufactured by NimbleGen Systems Limited (Madison, WI), containing duplicate sets of three independent, non-identical 60-mer probes per gene model plus control probes and labeling controls for a total of 65,965 probe sets corresponding to 55,970 gene models predicted on the P. trichocarpa genome sequence version 1.0 and 9,995 aspen cDNA sequences (Populus tremula, Populus tremuloides, and P. tremula × P. tremuloides), was used for expression analysis. The Populus version 2.0 oligoarray is fully described in the platform Gene Expression Omnibus (GEO) at NCBI (http://www.ncbi.nlm.nih.gov/geo; GPL2699). Single dye labeling of samples and hybridization procedures were performed at the NimbleGen facilities (NimbleGen Systems, Reykjavik, Iceland) following their standard protocol. Expression data were processed to assure a high specificity in the following manner: (1) all independent 60-mer oligos for the 55,970 genes were blasted against the Populus genome v1.1 (Tuskan et al. 2006) available on http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html and only probes with less than 10% homology to gene models other than the gene model target were retained for the further analysis. Due to this stringent filtering, about 30% of the genes (16,667) were excluded since all three independent probes failed the given specificity and (2) fluorescence data were normalized using quantile normalization and Robust Multichip Average (Irizarry et al. 2003) implemented in the ARRAYSTAR software (DNASTAR, Madison, WI) and 3) A Student’s t test, with FDR (Benjamini-Hochberg) multiple testing corrections, was applied to the duplicate sets on the array and the three biological repetitions using ARRAYSTAR software. Transcripts with a significant p value (<0.05) and ≥5.0-fold change in transcript level were considered as differentially expressed between P. trichocarpa ECM and P. deltoides ECM. Average expression levels from the three biological repetitions were calculated for each gene and were used for the calculation of the expression ratio. The complete expression dataset is available as series (accession number GSE26416) at the Gene Expression Omnibus at NCBI (http://www.ncbi.nlm.nih.gov/geo/).
Results
Mycorrhizal colonization
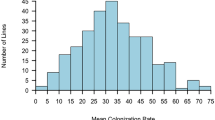
Fourteen weeks after inoculation, root systems of the 300 progeny and two parental genotypes were exclusively colonized by L. bicolor. No greenhouse inhabiting contaminant ectomycorrhizal fungi, such as Thelephora terrestris, were observed on the roots. Progeny and parental clones could be ranked based on their degree of mycorrhizal colonization (Fig. 1). The two parents greatly differed in their mycorrhizal colonization rate; P. trichocarpa exhibited a colonization rate of 45% ± 2, whereas P. deltoides showed a rate of 15% ± 3. The percentages of ECM colonization among the progeny varied from 8% (hybrid clone No. 661300230) to 88% (hybrid clone N°661300592), with an average of 47%. The genetic variation in this interspecific hybrid family was assessed by a linear mixed model (Table 1). ANOVA results showed a significant genotype and observer effects. No block effect was detected. The broad-sense heritability of the percentage of mycorrhizal colonization was 0.40 (CV G = 20.82%) before correction of the observer effect while after correction it was 0.38 (CV G = 22.94%).

Distribution of Populus deltoides (female) and Populus trichocarpa (male) and their F1 progeny following their root colonization by Laccaria bicolor. The bars represent the number of genotypes distributed in each class according to their percentages of mycorrhizal colonization. The percentage of P. deltoides is identified by the pink line while that of P. trichocarpa is identified by the blue line
QTL detection
A single QTL (Myc_d1 on linkage group LG II d), explaining 10.9% of the variation of the mycorrhizal colonization, was detected (LOD score = 1.4, significance = 0.018; Table 2) on the genetic map of P. deltoides (Jorge et al. 2005). On the genetic map of P. trichocarpa (Jorge et al. 2005), three QTLs—Myc_t1, Myc_t2m, and Myc_t3—respectively explaining 28.3%, 3.0%, and 1.8% of the mycorrhizal colonization trait, were detected on the respective linkage groups LG I t (LOD score = 4.0, significance = 0.002), LG III t (LOD score = 1.1, significance = 0.048), and LG XI t (LOD score = 1.1, significance = 0.024; Table 2). None of these QTLs co-localized with QTLs for partial resistance to Melampsora × columbiana or M. larici-populina (Tagu et al. 2005; Yin et al. 2004). The nearest flanking SSR and/or SNP markers allowed the positioning of these QTL regions onto the Populus genome v2.0 (Tuskan et al. 2006). These QTL intervals encompassed 15 Mb on the Populus chromosome II (Myc_d1), 4.2 Mb on the chromosome I (Myc_t1), 2.4 Mb on the chromosome III (Myc_t2), and 1.7 Mb on the chromosome XI (Myc_t3).
Analysis of microarrays transcript profiling
Among all genes, 2,034 myccorhiza-regulated transcripts corresponded to the QTL intervals, i.e., Myc_d1 (1528), Myc_t1 (264), and Myc_t2 (242). No significant transcripts were found for QTL Myc_t3. By considering a change in gene expression at a ≥5-fold difference between P. trichocarpa ECM and P. deltoides ECM, we found 210 transcripts associated with Myc_d1 (154), Myc_t1 (27), and Myc_t3 (29) out of a total of 8,546 differentially expressed across the entire genome. In a second step involving the four QTLs, we identified 41 transcripts which displayed a significant (p value ≤ 0.05) ≥5-fold gene expression among the 1,543 (p value ≤ 0.05; ≥5.0-fold) differentially expressed genes between the mycorrhizas of the two parents (Table 3). Five of the 41 transcripts were among the ten most differentially represented transcripts between P. trichocarpa ECM and P. deltoides ECM (Table 4). Of these 41 transcripts, 27 corresponded to genes of known function and 14 to genes of unknown function.
Discussion
The current study confirmed the results of Rosado et al. (1994) and Tagu et al. (2001; 2005), who showed that the ability to form ectomycorrhizas in Pinus elliotii and P. trichocarpa × P. deltoides, respectively, was partially under the genetic control of the host. Using 146 progenies from the 54B family, Tagu et al. (2001, 2005) found that the heritability of the mycorrhizal development ranged between 0.49 (Tagu et al. 2001) and 0.09 (Tagu et al. 2005). With a larger progeny (300 clones), our statistical analyses have shown a significant genotype effect on ectomycorrhizal formation and a broad-sense heritability of the percentage of mycorrhizal colonization of 0.38 (CV G = 22.94%), indicating moderate genetic control of the trait. Our results, as in previous studies (Tagu et al. 2001; 2005), indicate a large difference in the response of the two parents to L. bicolor colonization (45% for P. trichocarpa and 15% for P. deltoides). The mean of the progeny in our study was 47% (vs. 35% in Tagu et al. 2005) was not significantly different from that of the P. trichocarpa parent. We thus hypothesize that the genes involved in ECM colonization are dominant and inherited from the P. trichocarpa parent, just as the bimodality in the distribution might be explained by the dominant hypothesis (Fig. 1). Interestingly, 32% of the progeny, such as the clone 661300592, showed transgressive segregation and a higher percentage of mycorrhizal colonization than that of their parents. This result may be due to the phylogenetic distance that exists between the two parents, which functionally could lead to the formation of heterozygotes in the hybrid progeny and heterosis for this trait. If validated, this suggests that the ability to form ectomycorrhizas in Populus could be manipulated via breeding.
In the current study, a QTL analysis identified four QTLs, one on the maternal P. deltoides map and three on the paternal P. trichocarpa map (Jorge et al. 2005). Until a saturated, higher resolution genetic map of P. deltoides and P. trichocarpa can be created, there will remain a difficulty in narrowing the QTL intervals associated with ECM colonization while increase in the observed population would elicit also more recombinant in the genomic regions of interest. The genomic regions corresponding to the detected QTLs encompass several megabases. In order to identify candidate genes which may be playing a role in the mycorrhizal development, we selected genes differentially expressed between P. trichocarpa and P. deltoides ECM and located in the QTL genomic regions. Among the 8,546 and 1,543 (p value ≤ 0.05; ≥5.0-fold) differentially expressed genes in the entire genome between the mycorrhizas of the two parents, 41 (p value ≤ 0.05; ≥5.0-fold) transcripts were located in the QTL genomic regions: 20 in Myc_d1, 14 in Myc_t1, and seven in Myc_t2.
Among these 41 transcripts, 25 were overrepresented in P. deltoides and 16 were overrepresented in P. trichocarpa. Based on our inoculation results, we hypothesize that the genes whose transcripts are overrepresented in P. trichocarpa play a positive role in ectomycorrhizal development, while the genes whose transcripts are overrepresented in P. deltoides play a negative role. That is, the most overrepresented gene in P. trichocarpa mycorrhizas compared to P. deltoides mycorrhizas, found in the QTL Myc_t2 on the chromosome III, encodes for a protein, EREBP-4, an ethylene-responsive binding protein transcription factor (Leubner-Metzger et al. 1998; Ohta et al. 2000; Solano and Ecker 1998), known to play an important role in the regulation of pathogenesis related genes (Hao et al. 1998; Ohme-Takagi and Shinshi 1995). Moreover, Hirota et al. (2002) has shown that the auxin-regulated AP2/EREBP PUCHI gene, encoding a putative APETALA2/ethylene-responsive element binding protein transcription factor, is required for the coordinated pattern of cell divisions during lateral root formation in Arabidopsis thaliana. Finally, it is well-known that phytohormones are involved in ectomycorrhizal development (Barker et al. 1998; Slankis 1950; Gay et al. 1994; Karabaghli-Degron et al. 1998; Tagu et al. 2003). Rupp and Mudge (1985) showed that ethylene and auxin induce mycorrhiza-like roots on Pinus mugo. In the Eucalyptus globulus–Pisolithus microcarpus ectomycorrhiza, Tagu et al. (2003) showed that many plant genes are overexpressed during the mycorrhizal development and that the expression of the EgHypar gene, exhibiting homology with plant auxin-induced glutathione-S-transferases, was overexpressed not only in the ectomycorrhiza, but also by exogenous application of auxin. The upregulation of EREBP-4 in our study fits well with the previous knowledge on the involvement of hormonal interactions in ectomycorrhizal establishment.
Alternatively, microarray analysis in P. deltoides mycorrhizas compared to P. trichocarpa mycorrhizas detected transcripts for genes encoding proteins such as epoxide hydrolase, multi-antimicrobial extrusion proteins, AtKT1I proteins, WRKY proteins, and cytochrome p450, all reportedly playing roles in plant defenses (Gomi et al. 2003; Kiyosue et al. 1994; Li et al. 2008; Zou et al. 2004). Indeed, the epoxide hydrolase activity has been reported to be involved in the mechanisms of defense in plant–fungus interactions (Gomi et al. 2003). Epoxide hydrolase function in combination with the cytochrome p450 during the reticulation of cutin biopolymers in the cuticle layers of plant cell wall and are thought to create a physical barrier against pathogen attack (Blée and Schuber 1992; Hamberg and Fahlstadius 1992; Pinot et al. 1995). In addition to participation in the cutin biosynthesis, this enzyme produces epoxy fatty acids that may also act as antimicrobial compounds against fungal and bacterial pathogens (Kato et al. 1983, 1985; Masui et al. 1989; Ohta et al. 1990). Similarly, it is known that AtKT1I proteins inhibit the activity of extracellular proteases and can trigger both salicylic acid- and jasmonate/ethylene-dependent defense gene expression (Kariola et al. 2005; Li et al. 2004; Li et al. 2008; Ohme-Takagi and Shinshi 2000). Likewise, WRKY proteins are known to be induced in response to fungal elicitor and play an important role in the regulation of early defense–response genes (Eulgem et al. 1999; Zou et al. 2004). Thus, upregulation of this suite of genes in P. deltoides mycorrhizas may explain the lower mycorrhizal rates relative to P. trichocarpa. Indeed, the activation of defense mechanisms in P. deltoides may inhibit L. bicolor colonization, while in P. trichocarpa this inhibition may be repressed by the overexpression of EREBP-4 ethylene-sensitive transcription factor. Our results support the conclusions by Felten et al. (2010), Rupp et al. (1989), and Splivallo et al. (2009) that ethylene, released by Laccaria, plays a role in primary response of the host plant. Thus, the initial steps in successful colonization may involve Laccaria manipulating the host’s metabolic machinery and subsequently, decreasing their defenses.
There are limitations to the QTL approach presented in this study. A higher resolution QTL map is needed to narrow down the list of candidate genes. Validation of the detected QTLs different instead of alternate environments and alternate pedigrees is needed. The comparison of the gene expression between the mycorrhizas of the two parents integrates only a part of the interactions which take place during mycorrhizal establishment (i.e., early recognition and signal exchange, physical contact of the mycelium on the root, internal colonization, etc.). Nevertheless, the combination of QTL analysis and whole-genome microarray analysis does provide a list of candidate genes which appear to be involved into mycorrhizal that merit further investigation. In summation, based on our results, the interactions involved in mycorrhizal establishment and colonization are extremely complex and may not be as mutualistic as initially assumed.
References
Barker SJ, Tagu D, Delp G (1998) Regulation of root and fungal morphogenesis in mycorrhizal symbiosis. Plant Physiol 116:1201–1207
Blée E, Schuber F (1992) Occurrence of fatty acid epoxide hydrolases in soybean (Glycine max). Purification and characterization of the soluble form. Biochem J 282:711–714
Bradshaw HD, Stettler RF (1993) Molecular genetics of growth and development in Populus. I. Triploidy in hybrid poplars. Theor Appl Genet 86:301–307
Cornelius J (1994) Heritabilities and additive genetic coefficient of variation in forest trees. Can J For Res 24:372–379
Di Battista C, Selosse MA, Bouchard D, Stenström E, Le Tacon F (1996) Variations in symbiotic efficiency, phenotypic characters and ploidy level among different isolates of the ectomycorrhizal basidiomycete Laccaria bicolor strain S238. Mycol Res 100:1315–1324
Duponnois R, Garbaye J (1991) Mycorrhization helper bacteria associated with Douglas fir-Laccaria laccata symbiosis: effects in aseptic and glasshouse conditions. Ann Sci For 48:239–251
Eulgem T, Rushton PJ, Schmelzer E, Hahlbrock K, Somssich IE (1999) Early nuclear events in plant defense signaling: rapid gene activation by WRKY transcription factors. EMBO J 18:4689–4699
Faivre-Rampant P, Bastien C, Augustin S, Breton V, Delplanque A, Mourier MC, Kertadikara A, Laurans F, Lefèvre F, Lesage MC, Menard M, Pinon J, Saintagne C, Valadon A, Villar M, Prat D (1999) Locating genomic regions involved in pest resistance in poplars. Proceedings of the International Poplar Symposium II, IUFRO, Orléans, France pp31
Felten J, Legué V, Ditengou FA (2010) Lateral root stimulation in the early interaction between Arabidopsis thaliana and the ectomycorrhizal fungus Laccaria bicolor. Plant Signal Behav 5:1–4
Frey-Klett P, Pierrat JC, Garbaye J (1997) Location and survival of mycorrhiza helper Pseudomonas fluorescens during establishment of ectomycorrhizal symbiosis between Laccaria bicolor and Douglas-fir. Appl Environ Microbiol 63:139–144
Gay G, Normand L, Marmeisse R, Sotta B, Debaud JC (1994) Auxin overproducer mutants of Hebeloma cylindrosporum Romagnesi have increased mycorrhizal activity. New Phytol 128:645–657
Gomi K, Yamamato H, Akimitsu K (2003) Epoxide hydrolase: a mRNA induced by the fungal pathogen Alternaria alternata on rough lemon (Citrus jambhiri Lush). Plant Mol Biol 53:189–199
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross mapping strategy and RAPD markers. Genetics 137:1121–1137
Hamberg M, Fahlstadius P (1992) On the specificity of a fatty acid epoxygenase in broad bean (Vicia faba L.). Plant Physiol 99:987–995
Hao D, Ohme-Takagi M, Sarai A (1998) Unique mode of GCC box recognition by the DNA-binding domain of ethylene-responsive element-binding factor (ERF Domain) in plants. J Biol Chem 273:26857–26861
Hirota A, Kato T, Fukaki H, Aida M, Tasaka M (2002) The auxin-regulated AP2/EREBP gene PUCHI is required for morphogenesis in the early lateral root primordium of Arabidopsis. Plant Cell 19:2156–2168
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Speed, exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264
Jorge V, Dowkiw A, Faivre-Rampant P, Bastien C (2005) Genetic architecture of qualitative and quantitative Melampsora larici-populina leaf rust resistance in hybrid poplar: genetic mapping and QTL detection. New Phytol 167:113–127
Karabaghli-Degron C, Sotta B, Bonnet M, Gay G, Le Tacon F (1998) The auxin transport inhibitor 2, 3, 5-triiodobenzoic acid (TIBA) inhibits the stimulation of in vitro lateral root formation and the colonization of the tap-root cortex of Norway spruce (Picea abies) seedlings by the ectomycorrhizal fungus Laccaria bicolor. New Phytol 140(4):723–733
Kariola T, Brader G, Li J, Palva ET (2005) Chlorophyllase 1 a damage control enzyme affects the balance between defensepathways in plants. Plant Cell 17:282–294
Kato T, Yamaguchi Y, Uehara T, Yokoyama T, Namai T, Yamanaka S (1983) Defense mechanism of the rice plant against rice blast disease. Naturwissenschaften 70:200–201
Kato T, Yamaguchi Y, Abe N, Uehara T, Namai T, Kodama M, Shiobara Y (1985) Structure and synthesis of unsaturated trihydroxy C18 fatty acids in rice plants suffering from rice blast disease. Tetrahedron Lett 26:2357–2360
Kiyosue T, Beetham JK, Pinot F, Hammock BD, Yamaguchi-Shinozaki K, Shinozaki K (1994) Characterization of an Arabidopsis cDNA for a soluble epoxide hydrolase gene that is inducible by auxin and water stress. Plant J 6:259–269
Lander ES, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lefèvre F, Mc G-M, Faivre-Rampant P, Villar M (1998) A single gene cluster controls incompatibility and partial resistance to various Melampsora larici-populina races in hybrid poplars. Phytopathology 88:156–163
Leubner-Metzger G, Petruzzelli L, Waldvogel R, Vögeli-Lange R, Meins F (1998) Ethylene-responsive element binding protein (EREBP) expression and the transcriptional regulation of class I-1, 3-glucanase during tobacco seed germination. Plant Mol Biol 38:785–795
Li J, Brader G, Palva ET (2004) The WRKY70 transcription factor: a node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell 2004(16):319–331
Li J, Bradera G, Tapio Palva E (2008) Kunitz trypsin inhibitor: an antagonist of cell death triggered by phytopathogens and fumonisin B1 in Arabidopsis. Mol Plant 1:482–495
Martin F, Nehls U (2009) Harnessing ectomycorrhizal genomics for ecological insights. Cur Opin Plant Biol 12:508–515
Martin F, Duplessis S, Ditengou F, Lagrange H, Voiblet C, Lapeyrie F (2003) Developmental cross talking in the ectomycorrhizal symbiosis: signals and communication genes. New Phytol 151:145–154
Martin F, Kohler A, Duplessis S (2007) Living in harmony in the wood underground: ectomycorrhizal genomics. Cur Opin Plant Biol 10:204–210
Martin F, Aerts A, Ahrén D, Brun A, Duchaussoy F, Gibon J, Kohler A, Lindquist E, Pereda V, Salamov A, Shapiro HJ, Wuyts J, Blaudez D, Buée M, Brokstein P, Canbäck B, Cohen D, Courty PE, Coutinho PM, Danchin EGJ, Delaruelle C, Detter JC, Deveau A, DiFazio S, Duplessis S, Fraissinet-Tachet L, Lucic E, Frey-Klett P, Fourrey C, Feussner I, Gay G, Grimwood J, Hoegger PJ, Jain P, Kilaru S, Labbé J, Lin YC, Legué V, Le Tacon F, Marmeisse R, Melayah D, Montanini B, Muratet M, Nehls U, Niculita-Hirzel H, Oudot-Le Secq MP, Peter M, Quesneville H, Rajashekar B, Reich M, Rouhier N, Schmutz J, Yin T, Chalot M, Henrissat B, Kües U, Lucas S, Van de Peer Y, Podila G, Polle A, Pukkila PJ, Richardson PM, Rouzé P, Sanders IR, Stajich JE, Tunlid A, Tuskan G, Grigoriev IV (2008) Symbiosis insights from the genome of the mycorrhizal basidiomycete Laccaria bicolor. Nature 452:88–92
Masui H, Kondo T, Kojima M (1989) An antifungal compound 9,12,13-trihydroxy-(E)-10- octadecenoic acid from Colocasia antiquorum inoculated with Ceratocystis fimbriata. Phytochemistry 28:2613–2615
Nehls U, Mikolajewski S, Magel E, Hampp R (2001) Carbohydrate metabolism in ectomycorrhizas: gene expression, monosaccharide transport and metabolic control. New Phytol 150:533–541
Ohme-Takagi M, Shinshi H (1995) Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 7:173–182
Ohme-Takagi M, Shinshi H (2000) Three ethylene-responsive transcription factors in tobacco with distinct transactivation functions. Plant J 22:29–38
Ohta H, Shida K, Peng LY, Furusawa I, Shishiyama J, Aibara S, Morita Y (1990) The occurrence of lipid hydroperoxide-decomposing activities in rice and the relationship of such activities to the formation of antifungal substances. Plant CellPhysiol 31(8):1117–1122
Ohta M, Ohme-Takagi M, Shinshi H (2000) Three ethylene-responsive transcriptional factors in tobacco with distinct transactivation functions. Plant Cell 12:393–404
Pachlewski R, Pchlevska J (1974) Studies on symbiotic properties of mycorrhizal fungi of pine (Pinus sylvestris L.) with the aid of the method of mycorrhizal synthesis in pure cultures on agar. PhD Thesis. Forest Institute, Warsaw
Peter M, Courty PE, Kohler A, Delaruelle C, Martin D, Tagu D, Frey-Klett P, Duplessis S, Chalot M, Podila G, Martin F (2003) Analysis of expressed sequence tags from the ectomycorrhizal basidiomycete Laccaria bicolor and Pisolithus microcarpus. New Phytol 159:117–129
Peterson L, Bonfante P (1994) Comparative structure of vesicular-arbuscularmycorrhizas and ectomycorrhizas. In: Robson AD, Abbort LK, Malajczuk N (eds) Management of mycorrhizas in agriculture, horticulture and forestery. Kluwer, Dordrecht, pp 79–88
Pinot F, Grant DF, Beetham JK, Parker AG, Borhan B, Landt S, Jones AD, Hammock BD (1995) Molecular and biochemical evidence for the involvement of the Asp333-His523 pair in catalytic mechanism of soluble epoxide hydrolase. J Biol Chem 270: 7968–7974
Rosado SCS, Kropp BR, Piché Y (1994) Genetics of ectomycorrhizal symbiosis. I. Host plant variability and heritability of ectomycorrhizal root traits. New Phytol 126:105–110
Rupp LA, Mudge W (1985) Ethephon and auxin induce mycorrhiza-like changes in the morphology of root organ cultures of mugo pine. Physiol Plant 64:316–322
Rupp LA, DeVries HE, Mudge KW (1989) Effect of aminocyclopropane carboxylic acid and aminoethoxyvinylglycine on ethylene production by ectomycorrhizal fungi. Can J Bot 67:483–485
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Slankis V (1950) Effect of α-naphthalene-acetic acid on dichotomous branching of isolated roots of Pinus sylvestris. Physiol Plant 3:40–44
Smith SE, Reads DJ (eds) (1997) Mycorrhizal symbiosis. Academic, San Diego
Solano R, Ecker JR (1998) Ethylene gas: perception, signaling and response. Cur Opin Plant Biol 1:393–398
Splivallo R, Fischer U, Gobel C, Feussner I, Karlovsky P (2009) Truffles regulate plant root morphogenesis via the production of auxin and ethylene. Plant Physiol 150:218–229
Tagu D, Faivre-Rampant P, Lapeyrie Frey-klett P, Vion P, Villar M (2001) Variation in the ability to form ectomycorrhizas in the F1 progeny of an interspecific poplar (Populus spp.) cross. Mycorrhiza 10:237–240
Tagu D, Palin B, Balestrini R, Gelhaye E, Lapeyrie F, Jacquot JP, Sautière PE, Bonfante P, Martin F (2003) Characterization of a symbiosis- and auxin-regulated glutathione-S-transferase from Eucalyptus globulus roots. Plant Physiol Biochem 41:611–618
Tagu D, Bastien C, Faivre-Rampant P, Garbaye J, Vion P, Villar M, Martin F (2005) Genetic analysis of phenotypic variation for ectomycorrhiza formation an interspecific F1 poplar full-sib family. Mycorrhiza 15:87–91
Tuskan GA, Gunter LE, Yang ZMK, Yin TM, Sewell MM, DiFazio SP (2004) Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa. Can J For Res 34:85–93
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao BD, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Déjardin A, dePamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjärvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leplé JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouzé P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D (2006) The genome of black cottonwood, Populus trichocarpa. Science 313:1596–1604
Van der Schoot J, Pospiskova M, Vosman B, Smulders MJM (2000) Development and characterization of microsatellite markers in black poplar (Populus nigra L.). Theor Appl Genet 101:317–322
Villar M, Lefèvre F, Bradshaw HD, Teissier du Cros E (1996) Molecular genetics of rust resistance in poplars (Melampsora larici-populina Kleb/Populus sp.) by bulked segregant analysis in a 2 × 2 factorial mating design. Genetics 143:531–536
Yin TM, DiFazio SP, Gunter LE, Jawdy SS, Tuskan GA (2004) Mapping the rust resistant loci MXC3 and MER in Populus trichocarpa and assessing the intermarker linkage disequilibrium in the MXC3 region. New Phytol 164:95–106
Zou X, Seemann JR, Neuman D, Shen QJ (2004) A WRKY Gene from creosote bush encodes an activator of the abscisic acid signaling pathway. J Biol Chem 279:55770–55779
Acknowledgments
This project was supported by grants from the European Commission project ENERGYPOPLAR and EVOLTREE (to FM). JL was supported by a scholarship from the INRA, Region Lorraine, the ORNL, and the U.S. Department of Energy. Oak Ridge National Laboratory (ORNL) is managed by UT-Battelle, LLC for the US Department of Energy under contract no. DE-AC05-00OR2272. We thank Christine Delaruelle, Béatrice Palin, Judith Richter, Verónica Pereda, Saskia Reinhart, Anne Delaruelle, Simon Duchêne, Aurore Coince, and Jean-Louis Churin for their assistance in the inoculation and in measuring the colonization rates. We also thank Denis Tagu for his help and fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W. Boerjan
Rights and permissions
About this article
Cite this article
Labbé, J., Jorge, V., Kohler, A. et al. Identification of quantitative trait loci affecting ectomycorrhizal symbiosis in an interspecific F1 poplar cross and differential expression of genes in ectomycorrhizas of the two parents: Populus deltoides and Populus trichocarpa . Tree Genetics & Genomes 7, 617–627 (2011). https://doi.org/10.1007/s11295-010-0361-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-010-0361-3