
Abstract
Sucrose synthase is a key enzyme in sucrose metabolism in plant cells, and it is involved in the synthesis of cell wall cellulose. Although the sucrose synthase gene (SUS) family in the model plants Arabidopsis thaliana has been characterized, little is known about this gene family in trees. This study reports the identification of two novel SUS genes in the economically important poplar tree. These genes were expressed predominantly in mature xylem. Using molecular cloning and bioinformatics analysis of the Populus genome, we demonstrated that SUS is a multigene family with seven members that each exhibit distinct but partially overlapping expression patterns. Of particular interest, three SUS genes were preferentially expressed in the stem xylem, suggesting that poplar SUSs are involved in the formation of the secondary cell wall. Gene structural and phylogenetic analyses revealed that the Populus SUS family is composed of four main subgroups that arose before the separation of monocots and dicots. Phylogenetic analyses associated with the tissue- and organ-specific expression patterns. The high intraspecific nucleotide diversity of two SUS genes was detected in the natural population, and the π nonsyn/π syn ratio was significantly less than 1; therefore, SUS genes appear to be evolving in Populus, primarily under purifying selection. This is the first comprehensive study of the SUS gene family in woody plants; the analysis includes genome organization, gene structure, and phylogeny across land plant lineages, as well as expression profiling in Populus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Due to increasing global industrialization, increased human populations, and economic growth, the world faces very serious energy shortages and environmental pollution problems (Li et al. 2008). Biofuels are a potential alternative to crude oil, and their use has great potential to diminish the need for fossil fuels and thereby limit atmospheric CO2 accumulation. Ethanol generated from starch or sugar constitutes most of the biofuel that is currently produced, but this fuel will only meet a fraction of the global requirements. Thus, it has been suggested that large-scale use of fuel ethanol will require utilization of cellulosic biomass as a feedstock (Demirbas 2005). Forests contain most of the Earth’s terrestrial biomass, and thus large amounts of ethanol may be produced from the cellulose of wood cell. Among forest trees, the fast-growing poplar is emerging as a model for a woody cellulosic biofuel crop because it has a short rotation, high productivity in plantation forests, substantial carbon allocation to stems, and is easily planted. In addition to the productivity in plantation forests, the structure of cellulose polymers and chemical content of cellulose in wood cells is important for conversion into product ethanol. Because cellulose polymers are crystalline, they are highly recalcitrant to enzymatic breakdown for the formation of sugars that can be fermented to make ethanol. Therefore, a fundamental understanding of cellulose biosynthesis may help to enhance carbon sequestration and meet greater demands for biofuels.
Cellulose biosynthesis is a highly regulated process by which trees irreversibly sequester carbon in their primary and secondary cell walls. Sucrose, the main carbon source for cellulose synthesis, is transported to vegetative sink cells from photosynthetic source tissue cells, and its subsequent cleavage is the first step in photoassimilation in various metabolic pathways. The wood vascular tissue in the stem is comprised of highly active sink cells that use sucrose for rapid expansion and cellulose synthesis. In these cells, sucrose synthase (SUS; EC 2.4.1.13) is the major sucrolytic enzyme that catalyzes a reversible reaction that preferentially converts sucrose into fructose and UDP-glucose; the latter is the immediate precursor for cellulose biosynthesis (Joshi et al. 2004). SUS is a key player in regulating this influx of carbon into cellulose in stem vascular tissues (Hauch and Magel 1998). Consequently, a thorough knowledge of the expression patterns of SUS gene (SUS) families during tree development is required to accelerate our understanding of the role of SUS in regulating wood fiber properties and effectively manipulating cellulose biosynthesis.
Previous studies of the structure and function of SUS genes have focused mainly on model plants and important economic crops. In these species, multiple SUS isoforms that are encoded by several genes have been identified (Bieniawska et al. 2007; Hirose et al. 2008; Komatsu et al. 2002). For example, in the model plant Arabidopsis thaliana, the SUS family contains six distinct members grouped into three distinct AtSUS pairs based on their genomic structure, expression patterns, and phylogeny (Baud et al. 2004; Bieniawska et al. 2007). Rice (Oryza sativa) also has six SUS genes dispersed throughout the genome, of which four are expressed in a tissue- and stage-specific manner (Hirose et al. 2008). In particular, analysis of transgenic plants reveals that SUS members have specific functions in regulating cellulose deposition and cell wall formation during plant development. The repression of SUS by antisense RNA decreases the amount of cellulose in carrot (Daucus carota) (Tang and Strum 1999). Downregulation of SUS in cotton is also associated with reduced crystalline cellulose synthesis and repression of fiber cell initiation and elongation (Ruan et al. 2003). On the other hand, overexpression of corn SUS genes in the poplar not only significantly increases the cellulose content of secondary cell wall but also changes the cell wall ultrastructure in the stem, improving wood fiber quality for biofuel ethanol production (Coleman et al. 2009). Thus, members of the SUS family are divergent and differentially expressed and act as the limiting factor in cellulose biosynthesis during plant growth and development.
A comprehensive study of the structure, expression, and evolution of the SUS family in tree species is currently lacking. Consequently, the identification of new cDNAs and studies involving structure, expression profiling, and phylogeny of the SUS family from tree genomes is essential for improving wood fiber traits. Unlike model plant and crop species such as Arabidopsis and rice, most trees have long generation intervals and there is a shortage of mutant strains. Hence, it is very difficult to isolate genes by positional cloning or mutant library screening. As the genome of Populus trichocarpa is completely sequenced, the poplar is increasingly considered a model tree for genome-wide identification and characterization of gene families involved in growth and development (Tuskan et al. 2006).
We used poplar here as a model to address the significance of SUS multiplicity in trees. We report the identification and characterization of two previously unknown SUS genes, PtSUS1 and PtSUS2, from a mature xylem cDNA library of the economically important tree, Populus tomentosa. Using molecular cloning and bioinformatics analysis of the Populus genome, we demonstrate that poplar SUS is a multigene family comprised of at least seven members, each of which exhibits a different tissue-specific expression. We further show that the nucleotide diversity in PtSUS1 and PtSUS2 is high and that purifying selection has played an important role in the evolution of these genes.
Materials and methods
Plant materials
One-year-old P. trichocarpa (clone “Nisqually 1”) and P. tomentosa (clone “LM50”) trees were grown in a greenhouse under natural light conditions at Beijing Forestry University and were watered daily. Four-year-old P. tomentosa (clone “LM50”) trees were planted in the arboretum of P. tomentosa in Shandong province. For RNA extraction, fresh tissue samples of root, leaf, and apex were collected from the 1-year-old vegetatively propagated cuttings. The wood-forming tissues of upright stems, including phloem, cambium, developing xylem, and mature xylem, were collected as described (Zhang et al. 2010a). All tissues were immediately frozen in liquid nitrogen and stored at −80°C.
A panel of 40 unrelated individuals, representing almost the entire range of native P. tomentosa from the three regions (13 from the southern region, 13 from the northwestern region, and 14 from the northeastern region), was used to analyze the nucleotide diversity of PtSUS1 and PtSUS2 (“Electronic Supplementary Material”, Fig. S1).
Identification of PtSUS1 and PtSUS2 cDNAs
The P. tomentosa stem mature xylem cDNA library was constructed using the Superscript λ System (Life Technologies). The cDNA library was generated as part of our large-scale effort to identify genes expressed predominantly in the mature xylem of P. tomentosa stems. The constructed cDNA library consisted of 5.0 × 106 pfu with an insert size of 1.0–4.0 kb. Random end sequencing of 10,000 cDNA clones and comparison with all available Arabidopsis SUS sequences revealed 20 clones with high similarity to AtSUSs. Finally, two contigs were assembled from these EST sequences, which represented two full-length cDNAs named PtSUS1 and PtSUS2.
RNA extraction and SUS cDNA amplification
Total RNA was extracted from various tissues using the Plant Qiagen RNAeasy kit. Additional on-column DNase digestions were performed three times during the RNA purification using the RNase-Free DNase Set (Qiagen). RNA was then quantified and reverse transcribed into cDNA utilizing the SuperScript First-Strand Synthesis system and the supplied polythymine primers (Invitrogen). Gene-specific primers were designed for RT-PCR amplification of PtSUS1, PtSUS2, and PtrSUS1–7 and were based on the sequenced P. tomentosa ESTs and the P. trichocarpa genome sequence (“Electronic Supplementary Material”, Table S1).
Real-time quantitative PCR
Quantitative PCR was performed on a DNA Engine Opticon 2 machine (MJ Research) using the LightCycler-FastStar DNA master SYBR Green I kit (Roche). The PCR program included an initial denaturation at 94°C for 5 min, 40 cycles of 30 s at 94°C, 30 s at 58°C, and 30 s at 72°C, and a final melt curve of 70–95°C. The melting curve was used to check the specificity of the amplified fragments. All reactions were carried out in triplicate for technical and biological repetitions, respectively, and the real-time data were analyzed using the Opticon Monitor Analysis Software 3.1 tool. Specific primer sets were designed to target the 3′ untranslated region (UTR) for each gene using Primer Express 3.0 software (Applied Biosystems) (Table 1). The efficiency of the primer sets was calculated by performing real-time PCR on several dilutions of first-strand cDNAs. The efficiencies of the different primer sets were similar. The specificity of each primer set was checked by sequencing PCR products. The Actin primers were used as an internal control to confirm that equal quantities of total RNA were used as template for real-time PCR experiments. The results obtained for the different tissues we analyzed were standardized to the levels of Actin.
DNA extraction and SUS genomic DNA amplification
Total genomic DNA was extracted from fresh young leaves of each individual from the natural populations of P. tomentosa and genotype “Nisqually 1” of P. trichocarpa using the Plant DNeasy kit. The primer sets used for the amplification of SUSs were designed based on the sequenced cDNA of PtSUS1and PtSUS2 and the P. trichocarpa genome sequence (“Electronic Supplementary Material”, Table S2). PCR was performed in a final reaction volume of 25 μl containing 20 ng genomic DNA, 0.8 U Taq DNA polymerase (Promega), 50 ng forward primer, 50 ng reverse primer, 1× PCR buffer (Promega), and 0.2 mM each dNTPs (Promega). The PCR conditions were as follows: 96°C for 3 min, 30 cycles of 95°C denaturation for 30 s, 56°C annealing for 45 s, and 72°C extension for 1.5 min, with a final extension at 72°C for 5 min.
Subcloning and sequencing
All PCR or RT-PCR products were resolved by agarose gel electrophoresis, excised, and purified using Ultrafree®-DA (Millipore, Billerica, MA, USA) centrifugal filter units. The purified DNA was ligated into the pGEM®-T Easy Vector and transformed into JM109 competent cells (Promega). Plasmid DNA was isolated from overnight cultures using the QIAprep Spin Miniprep protocol (Qiagen). Plasmid DNA was sequenced on both strands using T7 and SP6 primers and the Big Dye Terminator version 3.1 Cycle Sequencing kit (Applied Biosystems) and a Licor 4300 genetic analyzer.
Phylogenetic analysis
The amino acid sequences of SUS families, including those of angiosperms, gymnosperms, and bacteria, were identified by searching public databases available at NCBI (http://www.ncbi.nlm.nih.gov) (“Electronic Supplementary Material”, Table S3). Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 4, and the neighbor-joining method was used to build phylogenetic trees (Tamura et al. 2007). Bootstrap analysis was performed using 1,000 replicates.
Nucleotide diversity analysis
Sequencher v.4.0 and BioEdit were used for sequence alignment, and manual editing was done to confirm sequence quality and remove primer sequences. Indels were excluded from all estimates. The DnaSP program version 4.90.1 (Rozas et al. 2003) was used to calculate summary statistics for polymorphisms and divergence. Nucleotide diversity was estimated using both the average number of pair-wise differences per site between sequences, π (Nei 1987), and the average number of segregating sites per site, θ w (Watterson 1975). Neutrality test statistics (Tajima 1989), D and F (Fu and Li 1993), were calculated and tested using 10,000 simulations to test the hypothesis that mutations in the gene were selectively neutral.
Results
Isolation of two distinct SUS cDNAs involved in xylem formation in P. tomentosa
The two full-length cDNAs (PtSUS1 and PtSUS2) encoding sucrose synthase were isolated from a cDNA library prepared from the mature xylem zone of P. tomentosa using RT-PCR. The PtSUS1 (2,703 bp; GenBank Accession No. GU559727) and PtSUS2 (2,762 bp; GU559728) clones had open reading frames encoding polypeptides of 805 and 803 amino acid residues, respectively, corresponding to ATG start codons at positions 107 and 129 bp and TGA and TAA stop codons at positions 2,524 and 2,540 bp (“Electronic Supplementary Material”, Fig. S2). The two cDNAs also contained 106 and 128 bp of 5′ UTR and 179 and 222 bp of 3′ UTR (“Electronic Supplementary Material”, Fig. S2), respectively. These two poplar SUS cDNAs shared 92.0% nucleotide sequence identity. Nucleotide sequences comparison of PtSUS1 and PtSUS2 with known full-length Arabidopsis SUS sequences indicated a maximum DNA sequence identity of 76.1% and 76.2% with AtSUS1 and AtSUS4, respectively. The predicted molecular masses of PtSUS1 and PtSUS2 were 92.3 and 92.0 kDa, respectively, which are approximately equivalent to those of the sucrose synthase subunits identified in other annual plants. The deduced amino acid sequences of PtSUS1 and PtSUS2 were 93.3% identical to each other. A BLASTP search using PtSUS1 and PtSUS2 as query sequences revealed an extensive homology (80.9–85.7%) with SUS of tomato, potato, and Arabidopsis (dicots) and considerable homology (74.1–80.0%) to corresponding sequences from wheat, rice, and maize (monocots). The PtSUS1 and PtSUS2 sequences showed many conserved motifs, similar to SUSs in other plants. The N-terminus of PtSUS1 and PtSUS2 contained the Ser11 and Ser9 phosphorylation sites that are conserved in most of the SUSs from different plants (“Electronic Supplementary Material”, Fig. S2). As reported for maize and potato, PtSUS1 and PtSUS2 had two transmembrane domains, i.e., residues 269–289 and 675–694 in PtSUS1 and residues 267–287 and 673–692 in PtSUS2. These data suggested that PtSUS1 and PtSUS2 are members of the SUS family.
Transcript profiling of PtSUS1 and PtSUS2 mRNAs in various poplar organs and tissues was examined using quantitative real-time RT-PCR and Actin as an internal control. PtSUS1 and PtSUS2 transcripts were present at different expression levels in the organs of the poplar, including the root, stem, and leaf (“Electronic Supplementary Material”, Fig. S3a). PtSUS1 and PtSUS2 were preferentially expressed in mature xylem tissue, with intermediate expression in root, developing xylem, and mature leaves (“Electronic Supplementary Material”, Fig. S3a). Interestingly, both transcripts appeared to be predominantly expressed in mature xylem sink tissue, consistent with their original identification as mature xylem specific cDNAs (“Electronic Supplementary Material”, Fig. S3a). Conversely, extremely low levels of PtSUS1 and PtSUS2 mRNA were detected in the primary tissues of the apex and cambium, and the levels of both mRNAs in secondary vascular tissues of the mature xylem were at least tenfold higher than in primary tissues. PtSUS2 mRNA was more abundant than PtSUS1 in all tissues examined, although they exhibited similar overall expression profiles (“Electronic Supplementary Material”, Fig. S3a). PtSUS2 was approximately tenfold more abundant in mature and developing xylem and about fivefold higher in leaves and roots than PtSUS1 (“Electronic Supplementary Material”, Fig. S3a). Thus, PtSUS1 and PtSUS2 are highly expressed in poplar and are differentially expressed in different tissues associated with wood formation.
Because PtSUS1 and PtSUS2 were predominantly expressed in the mature xylem tissues of poplar stems, we next analyzed growing xylem stem tissue in more detail: elongating stems of 4-year-old poplar trees were cut and marked longitudinally at six height points (0, 1.5, 3.0, 4.5, 6.0, and 7.5 m), and gene expression at each point was determined. PtSUS1 was most abundantly expressed at point 0 m of the basal stem, with the expression levels gradually decreasing towards the top of the stem (“Electronic Supplementary Material”, Fig. S3b). PtSUS2 was also most highly expressed at the bottom of the stem, but expression towards the stem top decreased to a lesser degree compared with PtSUS1 (“Electronic Supplementary Material”, Fig. S3b). Overall, PtSUS2 mRNA levels were consistently higher than those of PtSUS1 at all points in the mature xylem. PtSUS1 and PtSUS2 transcript abundance was in accordance with previous analysis of sucrose synthase activity of these SUSs in different heights of stem xylem in our lab (data not shown).
SUS belongs to a multigene family in Populus
To identify all the putative SUS genes in poplar, we searched the P. trichocarpa genome database with PtSUS1 and PtSUS2 sequences and detected seven distinct genes encoding full-length SUSs. The genomic and cDNA clones of all SUS family members in P. trichocarpa were then isolated, sequenced, and named PtrSUS1 through PtrSUS7 (Table 2). These seven genes were widely dispersed on four of the 19 Populus chromosomes and three scaffolds (Table 2). The predicted open reading frames ranged from 2,400 to 2,505 bp, and the corresponding deduced proteins varied from 800 to 835 residues (Table 2). The calculated molecular masses of the seven PtrSUSs ranged from 90.28 to 95.01 kDa, with PtrSUS5 being the largest (Table 2). All PtrSUS family members had a putative Ser phosphorylation site (Ser9 and Ser11) as well as the two characteristic sucrose synthase and glucosyl transferase domains (“Electronic Supplementary Material”, Fig. S4). Sequence comparison revealed that the genes share a high sequence homology at the nucleotide level (58.4% to 92.8% identity) within the coding region and at the amino acid level (54.0% to 92.9% identity) (Table 3). Three distinct PtrSUS pairs were found. A high sequence identity of nucleotide or amino acids was found between PtrSUS1 and PtrSUS2, PtrSUS4 and PtrSUS5, and PtrSUS6 and PtrSUS7, ranging from 87.6% to 92.9% (Table 3). PtrSUS3 was very different from the other six PtrSUSs (Table 3). To identify the evolutionary forces acting on poplar SUS genes, the non-synonymous/synonymous ratio (ω=d N/d S) was calculated. Within the coding regions, the average d N (0.22) was markedly lower than d S (0.66), and ω (0.33) was significantly less than 1, indicating that purifying selection was the dominant force driving the evolution of poplar sucrose synthase genes.
To learn how the intron/exon structure changed during evolution, the genomic and cDNA sequences of each PtrSUS member were compared, and the intron/exon organization of each gene was determined (Fig. 1). The number of introns in the transcriptional regions varied from 11 to 14, but the positions of the exon/intron boundaries in the coding region were relatively conserved among the seven PtrSUS genes. All introns started with 5′ G–T and ended with 3′ A–G and were in accordance with the GT–AG rule for a splice site. Based on the length and position of the introns, four patterns of intron–exon structures of the PtrSUS genes were revealed and designated I, II, III, and IV, respectively (Fig. 1). Pattern I in PtrSUS1 and PtrSUS2 contains a large 5′ intron comprising 1,893 and 2,674 bp at −5 bp upstream of the ATG initiation codon and 12 small introns with lengths varying from 81 to 249 bp within coding regions. Pattern II (PtrSUS3) includes 14 small introns with lengths ranging from 75 to 517 bp within coding regions (Fig. 1). Pattern III, which includes PtrSUS4 and PtrSUS5, has 11 small introns that vary in length from 73 to 400 bp (Fig. 1). Pattern IV (PtrSUS6 and PtrSUS7) has 12 small introns, ranging from 72 to 139 bp (Fig. 1). A comparison of the intron–exon structures in the coding regions revealed significant differences among these four PtrSUS patterns (Fig. 1). Pattern I had a fifth exon of 336 bp and an uninterrupted 11th exon of 564 bp. In pattern II (PtrSUS3), the 567-bp exon was split by an intron. Pattern III (PtrSUS4 and PtrSUS5) was characterized by a first exon of 225 bp and an eighth unsplit exon of 284 bp. In pattern IV (PtrSUS6 and PtrSUS7), the 13th exon was unsplit. These data indicated that PtrSUS gene structure is divergent in poplar.

Schematic representation of the structure of the SUS gene family in Populus. Exons and untranslated regions (UTRs) are indicated as black and blue boxes, respectively, and lines between boxes indicate introns
Evolution of SUS genes in angiosperms
To determine the evolutionary relationships among the SUS family members in poplar, a phylogenetic analysis was performed using the full-length protein sequences of the seven PtrSUSs. The phylogenetic dendrogram constructed by the neighbor-joining method formed two well-defined branches (Fig. 2). One branch contained PtrSUS1/2 and PtrSUS3, and the other branch contained PtrSUS4/5 and PtrSUS6/7. We further analyzed whether these phylogenetic subgroups corresponded to the PtrSUSs gene structure patterns defined in Fig. 1. Indeed the division of the PtrSUS family into four subgroups was consistent with the exon/intron organization patterns.

Phylogenetic relationships between poplar SUSs using Anabaena ASUS as an outgroup (refer to Table S3)
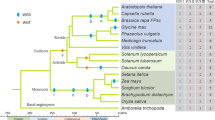
To explore the evolutionary relationship between poplar SUS genes and SUS genes in other plants, a rooted neighbor-joining phylogenetic tree of SUS proteins was constructed from 33 dicot sequences, 22 monocot sequences, five conifer sequences, and five bacteria sequences based on 1,000-replicate bootstrap values (Fig. 3). All bacterial SUS genes clustered into the same group (outgroup), whereas those from the land plants formed a monophyletic subfamily, showing that all plant SUS family members originated from a proteobacterial or ancestral type (Lunn 2002) (Fig. 3). Using five SUSs from bacteria as the outgroup, the plant SUS genes could be subdivided into four clearly distinct subgroups with high statistical support, including genes from angiosperms and gymnosperms. These subgroups were designated class I, class II, class III, and class IV, respectively (Fig. 3). Class III was represented only by sequences from gymnosperms, and the other three classes contained only angiosperms. The phylogenetic analysis suggested that the first gene duplication event that gave rise to SUS gene classes I–IV occurred before the divergence of angiosperms and gymnosperms. The robust resolution of the tree also enabled us to make further subdivisions within classes I, II, and IV. Each class was resolved into two branches, one specific for dicots and one for monocots. Thus, most second duplication events occurred before the monocot/dicot divergence. The nine poplar SUS genes—seven P. trichocarpa PtrSUSs and two P. tomentosa PtSUSs—were distributed in the dicot branch of classes I, II, and IV (Fig. 3). The PtrSUS4/5 and PtrSUS6/7 subclasses are more closely related to genes in Arabidopsis than they are to each other, suggesting that they originated from lineages that split off before Arabidopsis and poplar diverged (Fig. 3). The fact that several poplar gene pairs (PtrSUS1/2, PtSUS1/2) are clustered in the dicot branch of class I suggests that these genes arose by recent within-species duplications (Fig. 3).

Phylogenetic relationships between the poplar SUS genes and SUS genes in dicots, monocots, and conifers (refer to Table S3)
Transcript profiling in different tissues and organs
Using gene-specific primers (Table 1), real-time quantitative RT-PCR was used to detect the transcript profile for each member of the PtrSUS family in various tissues and organs of poplar. The specificity of each primer was verified by sequencing of the PCR products. Figure 4 illustrates the relative transcript levels of the seven PtrSUS genes normalized to the Actin expression level. PtrSUS family members were differentially expressed in the tissues and organs tested and exhibited four different expression patterns (Fig. 4). Both PtrSUS1 and PtrSUS2 were most abundantly expressed in the xylem tissue of stem and were moderately expressed in the mature leaf and root with similar expression patterns. However, PtrSUS2 was expressed at high levels in all tissues compared with PtrSUS1, and its expression level in xylem was tenfold higher than that of PtrSUS1. PtrSUS3 expression was significantly higher in mature leaf and apex compared with PtrSUS2 (Fig. 4). PtrSUS4 and PtrSUS5 were expressed most highly in mature leaf and root, whereas their expression was lowest in cambium, xylem, and young leaf (Fig. 4). PtrSUS6 and PtrSUS7 mRNA expression was largely confined to source mature leaf, but the transcripts were also detected at a lower level in root and apex (Fig. 4). Interestingly, transcript abundance for all PtrSUS members was higher in mature tissues of xylem and leaf than in young tissues. Thus, SUS genes in poplar were differentially expressed in different tissues, with PtrSUS1–3 being the predominant forms in the mature xylem zone (Fig. 4). The transcript profile of these seven PtrSUS genes appeared to be consistent with their phylogenetic relationships and the genomic structural patterns.

Relative mRNA abundance of seven PtrSUS family members in different tissues and organs
Intraspecific variation of PtSUS1 and PtSUS2 in the natural population
To characterize the intraspecific molecular evolution of the poplar SUS genes, the genomic sequence of PtSUS1 and PtSUS2 were obtained from 40 unrelated individuals, representing almost the entire naturally distributed range of P. tomentosa. The total length of the aligned sequence for each sample was approximately 10,996 bp, including 3,183 bp of the 5′ UTR with the 5′ intron, 4,830 bp of the coding region, 2,583 bp of introns, and 400 bp of the 3′ UTR (“Electronic Supplementary Material”, Table S4). Totally, there were 62 insertion/deletion (indels) polymorphisms found, which were all exclusively in the 5′ and 3′ UTRs or intron regions and ranged from 1 to 35 bp (average 5 bp) (“Electronic Supplementary Material”, Table S4). A more detailed analysis of nucleotide variation (excluding indels) was conducted over different regions of the PtSUS1 and PtSUS2 loci, and the profile of nucleotide diversity at these loci was plotted separately (Fig. 5). Different gene regions varied significantly with regard to nucleotide diversity (π), which ranged from 0.00051 (exon 1) to 0.03202 (3′ UTR) at the PtSUS1 locus and from 0.00000 (intron 7) to 0.03143 (intron 11) at the PtSUS2 locus. Across the samples, 504 SNPs were identified in these two genes, resulting in an average of about one SNP per 45.8 bp, 314 of which (62.3%) were considered common SNPs (frequency >0.10) (Table 4). Of the 504 unique sites, 172 were located in the exon regions of PtSUS1 and PtSUS2, of which 38, 39, and 95 variants occurred at the first, second, and third codon positions, respectively (Table 4). Among these, 90, 80, and two SNPs were categorized as silent, missense, and nonsense mutations, respectively (Table 4). The average π, as classified by gene structure in the 5′ UTR, exon regions, intron regions, and 3′ UTR, was 0.00925, 0.00562, 0.01279, and 0.03202 in PtSUS1, respectively, and 0.01914, 0.00605, 0.01279, and 0.00617 in PtSUS2, respectively. SNPs were less abundant in coding regions than in introns and flanking regions, suggesting that the coding region is conserved relative to the other regions under natural pressure both in PtSUS1 and PtSUS2. The PtSUS1 and PtSUS2 SNPs were distributed in a ratio of roughly 2.76:1 transitions to transversions (“Electronic Supplementary Material”, Table S5). The proportion of SNPs occurring at CpG dinucleotides was 20.2%, representing a 22-fold enrichment for polymorphisms at such relatively rare (~1%) but highly mutable positions. In general, both PtSUS1 and PtSUS2 displayed high nucleotide diversity, with π T = 0.00924 and θ w = 0.00949 for PtSUS1 and π T = 0.01093 and θ w = 0.01222 for PtSUS2 (Table 5). Within coding regions in PtSUS1 and PtSUS2, the nonsynonymous nucleotide substitutions (π nonsyn) was markedly lower than π syn, with the π nonsyn/π syn ratio ranging from 0.123 to 0.132, suggesting that diversity at the synonymous sites of exon regions resulted from strong purifying selection (Table 5).

The profile of nucleotide diversity at the PtSUS1 (a) and PtSUS2 (b) loci
Genetic differentiation within and among three geographically independent climatic regions was studied using the nucleotide diversity data from both PtSUS1 and PtSUS2 (Table 5). Levels of nucleotide variation (measured using π) in the three regions varied but showed similar patterns of π T, π sil, π syn, and π nonsyn (Table 5), suggesting that the level of selective constraint was similar between the climatic regions. We used Tajima's D (Tajima 1989) and Fu and Li's D (Fu and Li 1993) statistics to test whether a gene or genomic region is evolving randomly (neutral evolution) or whether the region is under selection (non-neutral evolution). For all three climatic regions, no significant departures from the neutral expectation were observed using Tajima's D (Table 5). Tajima's D was positive in each of the three climatic regions as opposed to the negative value found in the P. tomentosa population as a whole, suggesting that the three climatic regions have experienced a recent bottleneck or population contraction. Across the PtSUS1 and PtSUS2 loci, Fu and Li's D statistical tests were negative for the northwestern region and for the P. tomentosa population as a whole, revealing an excess of low-frequency polymorphisms in the species-wide samples and in the northwestern region (Table 6). Indeed in PtSUS1 and PtSUS2, 94 of 235 and 151 of 269 variants were singletons, accounting for 40.0% and 56.1% of the total segregation sites, respectively (Table 6).
The F ST statistic, a pairwise measure of population differentiation, was used to further assess the degree of climatic region-specific differentiation in both PtSUS1 and PtSUS2 (Table 6). Because of poplar outcrossing, wind pollination, and long-distance seed dispersal by wind, the level of population differentiation was expected to be generally low (Hamrick and Godt 1996). However, across the PtSUS1 and PtSUS2 loci, significant genetic differentiation was observed in the P. tomentosa population, with N ST and F ST ranging from 0.04560 and 0.04533 to 0.08606 and 0.08415, respectively (Table 6). Similarly, a significant genetic differentiation was noted between the northwestern and the other two regions; F ST ranged from 0.06246 between the northwestern and southern regions to 0.12686 between the northwestern and northeastern regions (Table 6). The highest number of shared polymorphisms (118 or 106) was found between the northeastern and southern regions, whereas the lowest number of shared polymorphisms (109 or 96) occurred between the northeastern and northwestern regions for both the PtSUS1 and PtSUS2 loci (Table 6). These observations are in agreement with F ST analyses in which the lowest genetic differentiation was found between northeastern and southern regions (F ST = 0.03560 or 0.03552) and the highest between northeastern and northwestern regions (F ST = 0.07481 or 0.12682).
Discussion
Structure and evolution of the SUS family in Populus
SUS genes are highly evolutionarily conserved, and members of this gene family are present both in gymnosperms and angiosperms. Unlike gymnosperms, angiosperm SUS families are, intriguingly, much more abundant and diverse. Multiple SUS isoforms have been identified both in dicot and monocot species. In our study, a primary goal was the thorough analysis of the structure and evolution of the SUS family in the model tree species, Populus. The overall gene structure profile in PtrSUSs was very similar to that of sucrose synthase genes isolated from Arabidopsis and rice (Bieniawska et al. 2007; Hirose et al. 2008). It appears that the plant SUS family structure is conserved among Populus, Arabidopsis, and rice, but lineage-specific genome-wide duplication events and subsequent chromosomal rearrangements have differentially shaped in poplar. For example, the position and length of the 5′ intron in pattern I genes differ substantially between Arabidopsis and poplar. In poplar, the 5′ intron both in PtrSUS1 and PtrSUS2 is located at the −5 bp position, upstream of the ATG initiation codon, whereas the position of the 5′ intron in the Arabidopsis counterparts AtSUS1 and AtSUS4 varied from −28 to −46 bp. The 5′ intron was much longer in poplar (range, 1893–2,674 bp) than in Arabidopsis (1,084–1,259). The fundamental regulatory roles of the 5′ intron in SUS genes for enhancing gene expression or directing tissue-specific expression have been demonstrated in cotton, maize, and potato (Clancy and Hannah 2002; Fu et al. 1995; Luo et al. 2005). The preliminary results obtained in our laboratory for PtSUS1 suggest that the 5′ intron may regulate specific expression in mature xylem (data not shown).
The analysis of the occurrence of SUS in complete genomes contributes to our knowledge of the origin and evolution of the plant sucrose synthase. Using the protein sequences from the limited SUS family members known at the time, previous studies of SUS evolution in Arabidopsis and citrus suggested the existence of three or four distinct SUS subgroups (Baud et al. 2004; Komatsu et al. 2002). As more plant genomes were sequenced, more SUS family members were discovered, and most species were found to have multiple SUS genes. In this study, we comprehensively analyzed the evolution of the SUS family across wide plant taxonomic scales, and phylogenetic analysis suggested that the poplar SUS genes arose from a series of gene duplications that have occurred throughout vascular plant evolution (Fig. 3). This is the first report of SUS family sequences from both gymnosperms and angiosperms. Anabaena spp. SUSs, which are phylogenetically located at the base of the land plant SUSs, suggest that all land plant sucrose synthase genes may have originated about two to three billion years ago from proteobacteria or ancestral types (Lunn 2002). The first gene duplication event that gave rise to SUS family classes I–IV occurred before the angiosperm/gymnosperm divergence about 300 Myr ago (Savard et al. 1994). A later duplication of SUSs within classes I, II, and IV among angiosperms must have arisen before the separation of the monocots and dicots, which is thought to have occurred about 200 Myr ago (Mitchell-Olds and Clauss 2002). Within class IV, the PtrSUS4/5 and PtrSUS6/7 subclasses are more closely related to genes from Arabidopsis than they are to each other, suggesting that the PtrSUS4/5 and PtrSUS6/7 lineages separated before the divergence of Arabidopsis and poplar 100–120 Myr ago (Tuskan et al. 2006). The PtrSUS1/2 and PtSUS1/2 subclasses appear to be the most recently diverged poplar within-species duplications and may have split after the divergence of the Populus and Salix lineages not more than 60–65 Myr ago (Tuskan et al. 2006).
SUS families are differentially expressed in Populus
The presence of duplicated SUS genes raises the question of their functional redundancy. As discussed above, the significant divergence found among the poplar SUS genes, including the 5′ UTR, suggested that these genes may differ in their expression levels or functions. In our study, the expression profile of each member of the seven poplar SUS families was investigated as a first step towards understanding the respective functions. The seven poplar SUS genes exhibited distinct but partially overlapping expression patterns (Fig. 4). Among the PtrSUS family members, four expression patterns were present in the organs and tissues studied (Fig. 4). PtrSUS1 and PtrSUS2 in expression pattern 1 were predominantly expressed in xylem tissues of stem, suggesting that they are associated with wood cell wall or cellulose biosynthesis. A growing body of evidence indicates that one of the major roles of SUS is to channel UDP glucose for the synthesis of cell wall polysaccharides such as cellulose in various plants (Haigler et al. 2001). The involvement of SUS in cellulose deposition is supported by the presence of PtrSUS1 and PtrSUS2 into class I in phylogenetic analysis with several previously reported SUS genes from cotton and pea in dicots and maize and wheat in monocots (Albrecht and Mustroph 2003; Barratt et al. 2001; Hardin et al. 2006; Ruan et al. 2003). Ruan et al. (2003) reported that a 70% reduction in SUS activity suppresses cell initiation and elongation in cotton fibers, probably by inhibiting cellulose biosynthesis in cell walls. The importance of SUS members in regulating wood formation is also illustrated by the overexpression of an exogenous corn SUS in the poplar tree, resulting in significantly increased secondary cell wall cellulose content and changes in cell wall ultrastructure in the tree stem (Coleman et al. 2009). However, a recent study of both the SUSs and invertases mutants proposed that cytosolic invertase, rather than sucrose synthase, is required for normal growth and cellulose biosynthesis in the cell wall of Arabidopsis (Barratt et al. 2009). Thus, systematic expression studies of each SUS member are still needed to obtain a complete overview for the entire family. Moreover, high transcript levels of PtrSUS2 compared to those of PtrSUS1 in most tissues were also observed for the corresponding SUS genes in Arabidopsis (Baud et al. 2004). In contrast, PtrSUS4/5 and PtrSUS6/7 clustered closely in class IV with Arabidopsis AtSUS5/6 and rice OsSUS5/6, which have been reported to be highly expressed in leaf (Baud et al. 2004; Hirose et al. 2008). The precise function of these two genes, however, remains unclear. Most closely related SUS genes have similar regulatory patterns, whereas more distant classes have less similar patterns (Fig. 4). Therefore, the poplar SUS members show tissue- and organ-specific expression, the patterns of which are congruent with the evolutionary relationships based on protein sequences.
Intraspecific diversity and differentiation of PtSUS genes in the natural population
Quantification of genetic variation in natural populations is key to understanding the forces responsible for shaping evolutionary change. The evolutionary context of population processes in P. tomentosa will be greatly enhanced by understanding patterns of genetic variation of nuclear genes in unrelated individuals in the native population, for which relatively few data are available (Zhang et al. 2007). In this study, we first comprehensively investigated the intraspecific nucleotide diversity of two full-length SUS genes in the natural population of P. tomentosa. A comparison of PtSUS1 and PtSUS2 with another nuclear gene PtCOBL4 detected in the same sample panel indicated that the total nucleotide diversity of PtSUS1 (π T = 0.00924) and PtSUS2 (π T = 0.01093) was slightly higher than that of PtCOBL4 (π T = 0.00502) (Zhang et al. 2010b) but significantly lower than Arabidopsis lyrata (π = 0.0225) (Ross-Ibarra et al. 2008). Compared with other species within genus Populus, the nucleotide diversity at the PtSUS loci in our study was notably higher than that in Populus tremula (π = 0.0042) (Ingvarsson 2008), Populus balsamifera (π = 0.0025) (Breen et al. 2009), and Populus nigra (π = 0.00702) (Chu et al. 2009). This discrepancy in average nucleotide diversity estimates across different poplar species under the same genus may be caused by a combination of factors such as differences in individual sampling strategies, the portion of the genome considered, demographic history, and differences in genetic backgrounds and mutation rates. Levels of average nucleotide diversity in coding regions were substantially lower than in the 5′ UTR, intron regions, and the 3′ UTR both at PtSUS1 and PtSUS2 loci, revealing that the coding regions are conserved relative to the other regions under natural pressure. Within coding regions, the π nonsyn/π syn ratio was significantly less than 1 for both genes (0.123 for PtSUS1 and 0.132 for PtSUS2), reflecting a strong purifying selection against substitutions that would result in amino acid replacements (Table 5).
Both PtSUS1 and PtSUS2 showed significant genetic differentiation in the native population of P. tomentosa, although low or moderate values of N ST (0.04560–0.08472) and F ST (0.04533–0.08415) were observed (Table 6). The level of population differentiation in our study was similar to that reported for five loci in the white poplar P. tremula (F ST range, 0.040–0.161) (Ingvarsson 2005) and for three loci from a Tacamahaca Spach poplar P. balsamifera (F ST range, 0.018–0.256) (Breen et al. 2009). Our study further demonstrated that the climatic regional differences appeared between the northwestern and each of the northeastern and southern regions of China. This result suggests that environmental factors probably play a significant role in the formation of genetic differentiation in native populations of Chinese P. tomentosa. Topography and climatic conditions in the regions naturally covered by P. tomentosa are the most obvious environmental differences between the three regions we studied (Huang 1992). The northeastern region consists of the city of Beijing and the provinces of Hebei and Shandong and is characterized by broad plains and an elevation of only 66 m, whereas the northwestern region, including provinces Shanxi, Shaanxi, and Gansu, is hilly/mountainous with a relatively high elevation (1,098 m). However, in comparison to the northeastern and northwestern regions, the southern region, which consists of the provinces Henan, Anhui, and Jiangsu, has obvious climatic differences such as high average temperatures and high annual average rainfall (Huang 1992). Consequently, the climatically and geographically disparate P. tomentosa isolates may have adapted to different temperatures and humidity levels as well as to other factors that correspond to latitudinal differences.
In conclusion, in this study, we explored the gene structure, phylogeny, and expression profiling of the entire sucrose synthase gene (SUS) family in a model tree, Populus. We demonstrate that SUS is a multigene family comprised of seven members, three of which were preferentially expressed in the stem xylem, suggesting that poplar SUSs are involved in the formation of secondary cell walls. Our analysis of the evolution of the SUS gene family in almost all plant lineages suggests that Populus SUS genes arose from a series of gene duplications. We found that the coding regions of SUS members are very highly conserved and that the synonymous sites of exon regions have undergone strong purifying selection during SUS evolution in Populus. Our findings provide an important insight into the mechanisms of the evolution of gene function and may help genetic engineers to regulate cellulose biosynthesis in trees for the sustainable production of wood biomass in the future.
References
Albrecht G, Mustroph A (2003) Localization of sucrose synthase in wheat roots: increased in situ activity of sucrose synthase correlates with cell wall thickening by cellulose deposition under hypoxia. Planta 217:252–260
Barratt DHP, Barber L, Kruger NJ, Smith AM, Wang TL, Martin C (2001) Multiple, distinct isoforms of sucrose synthase in pea. Plant Physiol 127:655–664
Barratt DHP, Derbyshire P, Findlay K, Pike M, Wellner N, Lunn J, Feil R, Simpson C, Maule AJ, Smith AM (2009) Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc Natl Acad Sci USA 106:13124–13129
Baud S, Vaultier MN, Rochat C (2004) Structure and expression profile of the sucrose synthase multigene family in Arabidopsis. J Exp Bot 55:397–409
Bieniawska Z, Barratt DHP, Garlick AP, Thole V, Kruger NJ, Martin C, Zrenner R, Smith AM (2007) Analysis of the sucrose synthase gene family in Arabidopsis. Plant J 49:810–828
Breen AL, Glenn E, Yeager A, Olson MS (2009) Nucleotide diversity among natural populations of a North American poplar (Populus balsamifera, Salicaceae). New Phytol 182:763–773
Chu YG, Su XH, Huang QJ, Zhang XH (2009) Patterns of DNA sequence variation at candidate gene loci in black poplar (Populus nigra L.) as revealed by single nucleotide polymorphisms. Genetica 137:141–150
Clancy M, Hannah LC (2002) Splicing of the maize Sh1 first intron is essential for enhancement of gene expression, and a T-rich motif increases expression without affecting splicing. Plant Physiol 130:918–929
Coleman HD, Yan J, Mansfield SD (2009) Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc Natl Acad Sci USA 106:13118–13123
Demirbas A (2005) Bioethanol from cellulosic materials: a renewable motor fuel from biomass. Energy Sources 27:327–337
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Fu HY, Kim SY, Park WD (1995) A potato Sus3 sucrose synthase gene contains a context-dependent 3′ element and a leader intron with both positive and negative tissue-specific effects. Plant Cell 7:1395–1403
Haigler CH, Ivanova-Datcheva M, Hogan PS, Salnikov VV, Hwang S, Martin K, Delmer DP (2001) Carbon partitioning to cellulose synthesis. Plant Mol Biol 47:29–51
Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Phil Trans R Soc Lond B 351:1291–1298
Hardin SC, Duncan KA, Huber SC (2006) Determination of structural requirements and probable regulatory effectors for membrane association of maize sucrose synthase 1. Plant Physiol 141:1106–1119
Hauch S, Magel E (1998) Extractable activities and protein content of sucrose phosphate synthase, sucrose synthase and neutral invertase in trunk tissues of Robinia pseudoacacia L. are related to cambial wood production and heart wood formation. Planta 207:266–274
Hirose T, Scofield GN, Terao T (2008) An expression analysis profile for the entire sucrose synthase gene family in rice. Plant Sci 174:534–543
Huang ZH (1992) The study on the climatic regionalization of the distributional region of Populus tomentosa. Journal of Beijing Forestry University 14:26–32
Ingvarsson PK (2005) Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 169:945–953
Ingvarsson PK (2008) Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics 180:329–340
Joshi CP, Bhandari S, Ranjan P et al (2004) Genomics of cellulose biosynthesis in poplars. New Phytol 164:53–61
Komatsu OA, Moriguchi T, Koyama K, Omura M, Akihama T (2002) Analysis of sucrose synthase genes in citrus suggests different roles and phylogenetic relationships. J Exp Bot 53:61–71
Li X, Weng JK, Chapple C (2008) Improvement of biomass through lignin modification. Plant J 54:569–581
Lunn JE (2002) Evolution of sucrose synthesis. Plant Physiol 128:1490–1500
Luo XY, Xiao YH, Li DM, Luo M, Hou L, Li XB, Yang X, Pei Y (2005) Cloning and analysis of the upstream regulated sequence of a cotton fiber sucrose synthase gene. Chin J Biochem Mol Biol 21:335–340
Mitchell-Olds T, Clauss MJ (2002) Plant evolutionary genomics. Curr Opin Plant Biol 5:74–79
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Ross-Ibarra J, Wright SI, Foxe JP, Kawabe A, DeRose-Wilson L, Gos G, Charlesworth D, Gaut BS (2008) Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata. PLoSONE3 6:e2411. doi:10.1371/journal.pone.0002411
Rozas J, Sánchez-Delbarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Ruan YL, Llewellyn DJ, Furbank RT (2003) Suppression of sucrose synthase gene expression represses cotton fiber cell initiation, elongation, and seed development. Plant Cell 15:952–964
Savard L, Li P, Strauss SH, Chase MW, Michaud M, Bousquet J (1994) Chloroplast and nuclear gene sequences indicate late Pennsylvanian time for the last common ancestor of extant seed plants. Proc Natl Acad Sci USA 91:5163–5167
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tang GQ, Strum A (1999) Antisense repression of sucrose synthase in carrot (Daucus carota L.) affects growth rather than sucrose partitioning. Plant Mol Biol 41:465–479
Tuskan GA, DiFazio S, Jansson S et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:188–193
Zhang DQ, Zhang ZY, Yang K (2007) Identification of AFLP markers associated with embryonic root development in Populus tomentosa. Silvae Genet 56:27–32
Zhang DQ, Du QZ, Xu BH, Zhang ZY, Li B (2010a) The actin multigene family in Populus: organization, expression and phylogenetic analysis. Mol Genet Genomics 284:105–119
Zhang DQ, Yang XH, Zhang ZY, Li B (2010b) Expression and nucleotide diversity of the poplar COBL gene. Tree Genet Genomes 6:331–344
Acknowledgments
This work was supported by grants from the Fundamental Research Funds for the Central Universities (No. BLJC200906), the project of National Natural Science Foundation of China (No. 30600479, 30872042), Major Science Foundation of Ministry of Education of China (No. 307006), Foundation for the Author of National Excellent Doctoral Dissertation of PR China (No. 200770), Program for New Century Excellent Talents in University (No. NCET-07-0084), and Introduction of Foreign Advanced Agricultural Science and Technology into China (No. 2009-4-22).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by W. Boerjan
Sequence data from this article have been deposited with the GenBank Data Library under accession numbers GU559727–GU559735 (PtSUS1, PtSUS2, PtrSUS1 to PtrSUS7 cDNA) and HM026865–HM026944 for the population sequences of PtSUS1 and PtSUS2 genes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Geographic location of each individual P. tomentosa tree used for nucleotide diversity analysis. The relevant Chinese provinces are shown. Each tree is indicated by a red dot (DOC 1411 kb)
Fig. S2
Nucleotide and deduced amino acid sequences of PtSUS1 (a) and PtSUS2 (b). Numbers on the left refer to the positions of nucleotides or amino acid residues (DOC 52 kb)
Fig. S3
Spatiotemporal expression patterns of PtSUS1 and PtSUS2 in poplar. a Relative transcript levels of PtSUS1 and PtSUS2 in various poplar tissues. b The spatial expression of PtSUS1 and PtSUS2 in the mature xylem of poplar (DOC 29 kb)
Fig. S4
Protein sequence alignment of PtrSUS family members. The numbers on the left are the positions of the amino acids in each protein, with gaps (dashes) included to maximize alignments. Identical and similar amino acids are shaded in pink and blue, respectively (DOC 189 kb)
Table S1
Primers used for sucrose synthase cDNA amplification (DOC 30 kb)
Table S2
Primers used for sucrose synthase gene amplification (DOC 42 kb)
Table S3
List of sucrose synthase gene sequences used in this study (DOC 59 kb)
Table S4
Lengths of gene regions analyzed in this study (DOC 36 kb)
Table S5
The frequencies of nucleotide substitution types in PtSUS1 and PtSUS2 (DOC 35 kb)
Rights and permissions
About this article
Cite this article
Zhang, D., Xu, B., Yang, X. et al. The sucrose synthase gene family in Populus: structure, expression, and evolution. Tree Genetics & Genomes 7, 443–456 (2011). https://doi.org/10.1007/s11295-010-0346-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-010-0346-2