
Abstract
The genus Prunus contains the subgenus Prunus incorporating the European plums (section Prunus), the North American plums (section Prunocerasus) and the apricots (section Armeniaca). In section Prunus, there are approximately 20 species, which occur in three levels of ploidy, diploid \( \left( {2n = 2x = 16} \right) \), tetraploid \( \left( {2n = 4x = 32} \right) \) and hexaploid \( \left( {2n = 6x = 48} \right) \). Despite a clear distinction between section Prunus and the other sections, phylogenetic relationships between species within the section are unclear. We performed a phylogenetic analysis on members of the section Prunus and three outgroup species using sequence data from four single-copy phylogenetically informative chloroplast DNA regions (atpB-rbcL, matK, rpl16, and trnL-trnF). After alignment, the analysed regions totalled 4,696 bp of sequence, containing 68 parsimony-informative sites and 14 parsimony-informative indels. Data were analysed using both maximum parsimony and Bayesian likelihood and phylogenetic trees were reconstructed. The analyses recovered trees with congruent topologies and similar levels of statistical support for relationships between taxa. They confirmed that species belonging to section Prunus form a monophyletic clade within Prunus. The section is resolved into four well-supported clades, which correspond to the geographical distribution of the species. The hexaploid species could not be resolved into distinct species clades but formed a well-supported group separate from the tetraploid species, highlighting the distinct evolutionary origins of the different polyploid groups. The close relationship between the hexaploids and Prunus divaricata, Prunus cerasifera and Prunus ursina indicates the former may have derived from an ancestor of P. cerasifera and its allies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Within the stone fruit genus Prunus L., the subgenus Prunus (classified as subgenus Prunophera by Rehder 1940) includes species of both plum and apricot from the Northern hemisphere. According to Krüssmann (1978), the subgenus comprises three sections: sect. Prunus, comprising plum species from Europe, Asia and North Africa; sect. Prunocerasus Koehne, which encompasses the North American plums; and sect. Armeniaca (Mill.) K. Koch, the apricots. The species in section Prunus can be distinguished from those of the other sections because they bear convolute leaves in the bud stage, glabrous ovaries and fruits, and pedunculate flowers. Species from section Prunocerasus can be distinguished from Prunus as they bear conduplicate leaves in the bud stage. In contrast, the section Armeniaca is distinguished by pubescent ovaries and fruits, flowers sessile or shortly pedunculate and leaves rolled up in the bud stage. In section Prunus, there are approximately 20 European and Asian plum species, several of which are of economic importance; most are diploids \( \left( {2n = 2x = 16} \right) \) and a few are tetraploids \( \left( {2n = 4x = 32} \right) \) or hexaploids \( \left( {2n = 6x = 48} \right) \). Although their phylogenetic differentiation from other sections is clear (Bortiri et al. 2001; Lee and Wen 2001; Shaw and Small 2004), the relationships amongst taxa within this group are not well understood.
Some plum species are thought to have far-eastern origins. Prunus salicina Lindl., reported to grow wild or to have been naturalised in Northern and South Eastern China (Kovalev 1941; Krüssmann 1978), is thought to have originated in the Yangtze River basin (Faust and Surányi 1999). This species is one of the most important plum species in cultivation today, and many new plum varieties have been obtained by hybridisation between P. salicina and other diploid Prunus species (Bellini et al. 1998; Boonprakob et al. 2001). In contrast, Prunus ussuriensis Kovalev & Kostina, endemic to North East China and East Siberia, and Prunus sogdiana Vassilcz., which is found in Tien Shan, Central Asia, grow wild in the forests of those regions (Sumnievich 1955; Paulov 1966). However, cultivars of both species are grown in Central Asia and the Russian Far East.
Further west, the cherry plum, Prunus cerasifera Ehrh., is widely cultivated in the Caucasus, Western Asia and Europe, mainly as a rootstock or as an ornamental, but it is also appreciated for its fruits (Eremin 1990). It is commonly thought (Browizc 1972; Kovalev 1941) that its wild form is the West-Asian species Prunus divaricata Ledeb. In addition, Prunus ursina Kotschy, which is found wild in the Levant, is sometimes regarded as a subspecies of P. divaricata (Browizc 1972), but it has also been proposed as a form of Prunus cocomilia Tenore (Dönmez and Yildirimli 2000). P. cocomilia is a small tree growing wild across Southern Italy, the Balkans and the mountains of Western Turkey as an Eastern Mediterranean endemic (Browizc 1972; Pignatti 1982). Another isolated European species, Prunus brigantina Vill., which grows wild in only a few Alpine valleys around Briançon, between France and Italy, has long been regarded as an apricot species instead of a plum (Pignatti 1982), despite its glabrous ovaries. However, recent molecular evidence has shown that it is not closely related to the apricot group (Hagen et al. 2001, 2002). The other plum species endemic to Europe, Prunus ramburii Boiss., is a thorny shrub that grows wild in the southern Spanish mountains. It is morphologically very similar to the widely distributed tetraploid Prunus spinosa but with even smaller leaves and fruits (Blanca and Diaz 1998) and is thought to be a relict species.
The hexaploid species Prunus domestica L., one of the most widely cultivated plums, has never been found in the wild and its origins are still the subject of controversy. Some authors (Crane and Lawrence 1931, 1938; Rybin 1936; Watkins 1981; Zeven and De Wet 1982) have regarded P. domestica as an allopolyploid hybrid species between a diploid cherry plum, P. cerasifera Ehrh., and the tetraploid blackthorn, P. spinosa L., which grows in Europe and West Asia. However, the participation of P. spinosa in the genesis of P. domestica has been questioned in recent times and it has also been proposed that P. domestica evolved from a hexaploid form of P. cerasifera (Zohary 1992). In addition, Prunus insititia L. has often been regarded as a subspecies of P. domestica (Bailey 1925; Browizc 1972; Pignatti 1982), or even as the same taxon (Woldring 2000).
Recently, a number of phylogenetic studies of Prunus have been undertaken, but these have been concerned with elucidating relationships between sections of the genus and have included only a limited number of taxa from section Prunus (Bortiri et al. 2001, 2002; Lee and Wen 2001; Shaw and Small 2004). These works involved sequencing analysis of both nuclear (S6pdh, ITS) and chloroplast (trnL-trnF) DNA regions. Furthermore, similar studies have analysed other genera within the Rosaceae family, such as Malus (Forte et al. 2001) and Fragaria (Potter et al. 2000), and even representatives of the whole family (Potter et al. 2002), by means of chloroplast sequence analysis (matK and trnL-trnF). In contrast to nuclear DNA, within which genes are present in multiple, often paralogous copies, the maternally inherited chloroplast genome provides convenient information for phylogenetic analyses of maternal lineages in taxonomic groups that contain different levels of ploidy between their member species.
In this study, we have investigated the phylogenetic relationships among Eurasian plum species, both cultivated and wild, belonging to section Prunus, by means of DNA sequence analysis of four phylogenetically informative regions of the chloroplast genome. The analysis clearly resolved well-supported relationships between all species investigated that were correlated to geographical origin, and that allowed inferences to be made about the evolutionary origins of the economically important domesticated plum species.
Materials and methods
Sampling
A total of 32 accessions were investigated. Twenty-six accessions, representing a total of 12 species and the three levels of ploidy (2x, 4x, 6x) (Zohary 1992; Watkins 1981) belonging to section Prunus, were sampled. For each of the nine diploid and one tetraploid species, two accessions were selected and analysed, and for the two hexaploid species, P. domestica and P. insititia, three accessions were used. In addition, two accessions each of the species Prunus maritima Wangenh. (sect. Prunocerasus Koehne), Prunus tomentosa Thunb. (subgenus Cerasus sect. Microcerasus Webb) and Prunus armeniaca L. (sect. Armeniaca (Mill.) K. Koch) were included as outgroup species. The majority of the material used was sourced from germplasm repositories. However, a number of accessions were collected from the wild. In order to check the species identity of the requested accessions, some morphological examination and cytometric analysis for ploidy determination was performed. The taxa investigated, along with their associated voucher specimen numbers, are listed in Table 1.
Amplification and sequencing of chloroplast DNA
Genomic DNA was extracted from fresh leaf tissue by the CTAB method (Doyle and Doyle 1987) and was diluted to a concentration of 10 ng µl−1 for use in polymerase chain reaction (PCR). Products were generated by PCR for each taxon for four regions of the chloroplast genome, none of which are located in the inverted repeat region, and thus are all single copy in the chloroplast genome (Table 2): atpB-rbcL region using primers atpB and rbcL (Chiang et al. 1998), matK using primers trnK685F and trnK2R (Hu et al. 2000), rpl16 using primers F71 and R1661 (Jordan et al. 1996) and trnL-trnF using primers c and f (Taberlet et al. 1991). Amplification of all PCR products was performed in a final volume of 100 μl comprising 2 µl template DNA, 1× PCR buffer, 2.0 mM Mg2+, 200 µM dNTPs, 0.2 µM each primer and 0.25 U Taq polymerase (Invitrogen). The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA), the purified products were sequenced from the primer pairs used for PCR using BigDye v3.1 (Applied Biosystems) chemistry according to the manufacturer’s specifications and sequence data were analysed on a semi-automated ABI 3100 capillary sequencer (Applied Biosystems). In the case of matK, internal primers matK4L (CTTCGCTACTGGGTGAAAGATG) and matK4R (CATCTTTCACCCAGTATCGAAG) were required for internal sequencing.
Sequence alignment and phylogenetic analysis
Forward and reverse sequences were assembled using SeqMan 4.06 (DNAStar Incorporated, USA) and aligned using MegAlign 4.06 (DNAStar Incorporated). Phylogenetically informative indels were coded as extra characters for use in the analysis of the data using parsimony. Alignments were analysed separately and then combined into a single alignment matrix containing data from all four chloroplast DNA regions. The alignments were imported into PAUP* 4.0b10 (Swofford 2003) and analysed using maximum parsimony as the optimality criterion (Swofford et al. 1996). A heuristic search employing 1,000 random addition sequence replicates with tree bisection and reconnection branch swapping was implemented and the strict consensus tree was then calculated from the most parsimonious trees. Phylogenetic bootstrapping was performed with 1,000 replicates to establish support for relationships inferred.
The general time-reversible model with ssgamma distribution rates (GTR+Г) was used for analysis of the combined alignment matrix using MrBayes 3.0b4 (Ronquist and Huelsenbeck 2003). Four incrementally heated Markov chains were run for 2 million generations, sampling every 20th generation, to produce 100,001 data points. Posterior probabilities were calculated from all trees produced after burn-in had been reached and the tree was visualised using PAUP* 4.0b10.
Results
Amplification and sequencing of chloroplast DNA
Single, discrete PCR products were generated with each of the primer pairs used for all taxa. ‘Ragged’ sequence ends were produced as a result of direct sequencing from PCR products and were removed from all sequences before alignment. Sequences were deposited in the European Molecular Biology Laboratory (EMBL) and accession numbers for all sequences produced are given in Table 3. The total sequence length for the 32 taxa investigated ranged from 4,561 to 4,612 bp and the total length of the aligned combined data matrix was 4,696 bp. The aligned matrix consisted of 4,604 invariable sites, 24 variable sites that were parsimony un-informative and 68 parsimony-informative sites. In addition, a total of 14 phylogenetically informative indels were coded for inclusion in the parsimony analysis.
Phylogenetic analysis
Separate analyses of the four chloroplast DNA regions all revealed similar phylogenetic relationships between the taxa investigated, but with lower resolution that the combined analysis (data not shown) and no conflicting hypotheses were revealed between the four separate analyses. The combined parsimony analysis recovered a single-most parsimonious tree with a tree length (L) = 111, consistency index (CI) = 0.98 and retention index (RI) = 0.97. The resultant tree is shown in Fig. 1 and relative bootstrap support values for relationships inferred are given above the branches. In the combined Bayesian likelihood analysis, a tree was constructed using MrBayes 3.0b4 from 99,001 trees sampled from generations after a stable likelihood had been reached, beginning at generation 20,001. Posterior probabilities for the Bayesian analysis were calculated from all post-burn-in generations and are presented on Fig. 2 above the branches. The topologies in both analyses were the same, except for an unsupported clustering of one accession of P. salicina with the two accessions of P. sogdiana in the parsimony analysis and thus both analyses were fully congruent except for the collapse of this node in the Bayesian analysis.

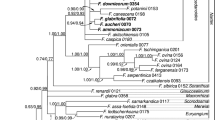
Strict consensus of the single-most parsimonious tree recovered by PAUP* 4.0b10 from the alignment of the atpB-rbcL, matK, rpl16 and trnL-trnF chloroplast DNA regions from 32 Prunus taxa. Tree statistics are tree length (L) = 111, consistency index (CI) = 0.98 and retention index (RI) = 0.97. Numbers above the branches are bootstrap values derived from 1,000 heuristic replicates. Clades defined A, B, C and D are discussed in the text

Majority-rules consensus phylogram of 99,001 trees generated from the Bayesian analysis of the total evidence matrix from the alignment of the atpB-rbcL, matK, rpl16 and trnL-trnF chloroplast DNA regions from 32 Prunus taxa using the GTR+Г model by MrBayes 3.0b4. Posterior probabilities are given above the branches and branch lengths are proportional to the mean estimates calculated under the GTR+Г model from the 99,001 trees used to construct the consensus tree
Relationships between the species of the section Prunus were clearly resolved in the phylogenetic analyses. In the parsimony analysis, the delimitation of P. brigantina, P. cocomilia, P. ramburii, P. spinosa, P. ursina and P. ussuriensis as distinct species was well supported (above 82% bootstrap support), whilst in the Bayesian analysis, support for these relationships and for the delimitation of P. cerasifera, P. divaricata and P. sogdiana as distinct species carried a posterior probability of 100%. The tetraploid species P. spinosa grouped in a well-supported clade along with P. brigantina and P. ramburii, whilst the two hexaploid species, P. domestica and P. insititia, grouped together with 100% posterior probability and 87% bootstrap support in a clade containing P. cerasifera, P. divaricata and P. ursina.
The section Prunus was supported as a distinct clade, separate from the outgroup species used. Within the section, four well-supported clades denoted A–D (Figs. 1 and 2) were recovered with 99% or greater posterior probability and, with the exception of clade D, 85% or greater bootstrap support.
Discussion
We have produced a robust phylogeny of the Prunus section Prunus using sequence data from the chloroplast atpB-rbcL, matK, rpl16 and trnL-trnF regions. The analysis provides clear evidence that the section Prunus is a well-supported monophyletic clade within Prunus as suggested by Rehder (1940) and Krüssmann (1978), and later concluded by Lee and Wen (2001) and Bortiri et al. (2001, 2002) according to molecular evidence within the context of genus Prunus. Analyses of chloroplast DNA in this section recovered a single-most parsimonious tree with strong bootstrap support and a well-resolved Bayesian likelihood analysis with significant posterior probabilities for four main clades within the section. Both analyses were congruent; however, some relationships carrying low bootstrap support (<70%) carried significant posterior probabilities in the Bayesian analysis.
The species P. salicina, P. sogdiana and P. ussuriensis are small trees with glabrous young twigs and conical calyces which extend through Central Asia, China, Japan and Siberia. They formed a monophyletic group within the section that was sister to all other species in the analysis. The two accessions of P. salicina studied, however, could not be resolved as a single species, possibly a reflection of the repeated domestication of this species throughout recent human history, during which interspecific hybridisation may have occurred more than once (Boonprakob et al. 2001), the earliest cultivar being known two millenia ago in China (Faust and Surányi 1999).
P. cocomilia, a species endemic to the Eastern sub-Mediterranean area, formed a distinct monophyletic clade B (Figs. 1 and 2), sister to all other European and West-Asian species, from which it can be morphologically distinguished, being a small tree with slightly thorny branches, glabrous pedicels and mucronate fruits. Two mountain relict species from West Europe, P. brigantina and P. ramburii, formed a distinct, well-supported monophyletic clade C (Figs. 1 and 2) with the tetraploid blackthorn, P. spinosa. The blackthorn is grouped within this clade C with P. ramburii with strong bootstrap support and posterior probability, suggesting that P. spinosa was formed from a diploid ancestor of P. ramburii as the result of a polyploidisation event. This is congruent with the previous findings (Mohanty et al. 2000, 2002) of higher haplotype diversity in South West Europe (Iberian Peninsula). This clade was sister to a larger clade D (Figs. 1 and 2) containing two subclades, D1 with P. divaricata, P. ursina and P. cerasifera, and D2 with the hexaploid species P. domestica and P insititia. P. divaricata and P. ursina, regarded by some authors as subspecies (Browizc 1972), cluster in a monophyletic clade D2 with the cultivated species P. cerasifera. The resolution between these species carried low bootstrap support; however, they were supported by 100% posterior probability as distinct species. Thus, there is evidence for a very close phylogenetic relationship between these morphologically very similar taxa.
By using chloroplast DNA for phylogenetic analyses, it is possible to reveal evolutionary relationships along the maternal line between species at different levels of ploidy. In this investigation, the hexaploid species P. domestica and P. insititia could not be resolved into distinct species clades, which is consistent with their proposed states as subspecies (Bailey 1925; Browizc 1972; Pignatti 1982). They formed a strongly supported subclade D1, sister to the abovementioned P. divaricata, P. cerasifera and P. ursina (99% posterior probability, 87% bootstrap support), from which they differ morphologically in having an obtuse base to the leaves, instead of acute, and no spines, which are sometimes found in branches of those diploids. Zohary (1992) suggested that the hexaploids were derived from P. cerasifera; the close relationship between the hexaploids and P. divaricata, P. cerasifera and P. ursina indicates the former may have derived from an ancestor of P. cerasifera and its allies. However, the hypothesis that P. domestica was formed from a hybridisation of P. cerasifera and P. spinosa could not be rejected in this analysis as sequence data were derived from chloroplast sequences, and therefore reflect only the maternal lineage of the species investigated. Furthermore, the possibility of other diploid species being involved as parental ancestors of subclade D1 could not be rejected.
In other recent studies of a section of Prunus, Shaw and Small (2004, 2005) dealt with section Prunocerasus. In their first paper, in which they analysed one example of each of the relevant species using several chloroplast sequences, the resulting cladogram bore little resemblance to groupings based on morphological characters. In the second paper, they looked at additional accessions of the various species for one sequence rpl16, and found that many species contained more than one of the three primary chloroplast DNA haplotypes. They attributed this sharing of chloroplasts to hybridisation and pointed out that a different choice of exemplars for the first paper could have resulted in a different inferred phylogeny. In our analysis of section Prunus, in which we used two or three accessions of each species, we found no such inconsistencies, except in the case of the two cultivated hexaploid species, which were not distinguishable on the basis of the sequence data used.
Concluding remarks
In this investigation, we have provided a phylogenetic evaluation of the Prunus section of the genus Prunus, incorporating all of the well-characterised species thought to belong to this section. Our analyses have shown that Prunus is a monophyletic section within the subgenus Prunus and that, despite being closely related and often morphologically similar, the recognised species within the section were, in general, distinguishable phylogenetically. The section is resolved into four well-supported clades (A–D), which correspond well to the geographical distribution of the species groups. The hexaploid plums formed a well-supported group D1 within clade D, separate from the tetraploid P. spinosa, which was a member of clade C, highlighting the distinct evolutionary origins of the different polyploid groups within the section and providing evidence for their closest diploid relatives. Despite the close relatedness of the species within section Prunus, analysis of the four chloroplast regions sequenced has provided a robust, well-supported phylogenetic framework for the section. An evaluation of the entire genus using these sequences could provide greater resolution in the other sections of Prunus and would place the findings presented here into a genus-wide context.
References
Bailey LH (1925) The standard encyclopedia of horticulture. Macmillan, New York
Bellini E, Giordani E, Nencetti V, Paffetti D (1998) Genetic relationships in Japanese plum cultivars by molecular markers. Acta Hortic 478:53–59
Blanca G, Díaz C (1998) Prunus L. In: Castroviejo S (ed) Flora Iberica vol. 6. Jardín Botánico de Madrid, Madrid, pp 444–450
Boonprakob U, Byrne DH, Graham CJ, Okie WR, Beckman T, Smith BR (2001) Genetic relationships among cultivated diploid plums and their progenitors as determined by RAPD markers. J Am Soc Hortic Sci 126:451–461
Bortiri E, Oh SH, Jiang J, Bagget S, Granger A, Weeks C, Buckingham M, Potter D, Parfitt D (2001) Phylogeny and systematics of Prunus (Rosaceae) as determined by sequence analysis of ITS and the chloroplast trnL-trnF spacer DNA. Syst Bot 26:797–807
Bortiri E, Oh SH, Gao FY, Potter D (2002) The phylogenetic utility of nucleotide sequence of sorbitol 6-phosphate dehydrogenase in Prunus (Rosaceae). Am J Bot 89:1697–1708
Browizc K (1972) Prunus L. In: Davis PH (ed) Flora of Turkey and the East Aegean islands, vol 4. Edinburgh University Press, Edinburgh, pp 8–12
Chiang TY, Schaal BA, Peng CI (1998) Universal primers for amplification and sequencing a noncoding spacer between the atpB and rbcL genes of chloroplast DNA. Bot Bull Acad Sin 39:245–249
Crane MB, Lawrence WJC (1931) Studies in sterility. Proceedings of the 9th International Horticultural Congress. London, pp. 100–116.
Crane MB, Lawrence WJC (1938) The genetics of garden plants. Macmillan, New York
Dönmez AA, Yildirimli S (2000) Taxonomy of the genus Prunus L. (Rosaceae) in Turkey. Turk J Bot 24:187–202
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Eremin GV (1990) New data on origin of Prunus domestica L. Acta Hortic 283:27–30
Faust M, Surányi D (1999) Origin and dissemination of plums. Hortic Rev 23:179–231
Forte AV, Dorochov DB, Savelyev NI. 2001. Phylogeny of wild Malus species revealed by morphology, RAPD markers, ITS1, 5.8rRNA, ITS2 and chloroplast gene matK sequences. Proceedings of 9th International Conference of Horticulture, vol. 1. Lednice, Czech Republic, 60–65
Hagen LS, Lambert P, Audergon JM, Khadari B (2001) Genetic relationships between apricot (Prunus armeniaca L.) and related species using AFLP markers. Acta Hortic 546:205–208
Hagen LS, Khadari B, Lamber P, Audergon JM (2002) Genetic diversity in apricot revealed by AFLP markers: species and cultivar comparisons. Theor Appl Genet 105:298–305
Hu JM, Lavin M, Wojciechowski MF, Sanderson MJ (2000) Phylogenetic systematics of the tribe Millettieae (Leguminosae) based on chloroplast trnK/matK sequences and ITS implications for evolutionary patterns in Papilionoideae. Am J Bot 87:418–430
Jordan WC, Courtney MW, Neigel J (1996) Low levels of intraspecific genetic variation at a rapidly evolving chloroplast DNA locus in North American duckweeds (Lemnaceae). Am J Bot 83:430–439
Kovalev NV (1941) Prunus L. In: Komarov VL (ed) Flora S.S.S.R, vol 10. Izd-vo Akademii nauk SSSR, St Petersburg, pp 510–521
Krüssmann G (1978) Manual of cultivated broad-leaved trees and shrubs, vol 3 Pru-Z. Timber Press, Portland
Lee S, Wen J (2001) A phylogenetic analysis of Prunus and the Amygdaloideae (Rosaceae) using ITS sequences of nuclear ribosomal DNA. Am J Bot 88:150–160
Maier RM, Neckermann K, Igloi GL, Kössel H (1995) Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251:614–628
Mohanty A, Martín JP, Aguinagalde I (2000) Chloroplast DNA diversity within and among populations of the allotetraploid Prunus spinosa. Theor Appl Genet 100:1304–1310
Mohanty A, Martín JP, Aguinagalde I (2002) Population genetic analysis of European Prunus spinosa using chloroplast DNA markers. Am J Bot 89:1223–1228
Pignatti S (1982) Flora d'Italia, vol 1. Edagricole, Bologna
Paulov NV (1966) Flora Kazakhstana. Kazakhstan Academy of Sciences, Alma Ata
Potter D, Luby JJ, Harrison RE (2000) Phylogenetic relationships among species of Fragaria (Rosaceae) inferred from non-coding nuclear and chloroplast DNA sequences. Syst Bot 25:337–348
Potter D, Gao F, Bortiri PE, Oh SH, Baggett S (2002) Phylogenetic relationship in Rosaceae inferred from chloroplast matK and trnL-trnF nucleotide sequence data. Plant Syst Evol 231:77–89
Rehder A (1940) A manual of cultivated trees and shrubs hardy in North America, 2nd edn. Macmillan, New York
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rybin WA (1936) Spontane und experimentell erzeugte Bastarde zwischen Schwarzdorn und Kirschpflaume und das Abstammungsproblem der Kulturpflaume. Planta 25:22–58
Shaw J, Small RL (2004) Addressing the hardest puzzle in American pomology: phylogeny of Prunus sect. Prunocerasus (Rosaceae) based on seven noncoding chloroplast DNA regions. Am J Bot 91:985–996
Sumnievich GP (1955) Flora Uzbekistana. Uzbekistan Academy of Sciences, Tashkent
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic inference. In: Hillis DM, Moritz C, Mable BK (eds) Molecular systematics, 2nd edn. Sinauer, Sunderland, pp 407–543
Swofford DL (2003) PAUP*. Phylogenetic analysis using parsimony (* and other methods), version 4.0b 10. Sinauer, Sunderland
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Watkins R (1981) Plums, apricots, almonds, peaches, cherries (genus Prunus). In: Hora B (ed) The Oxford encyclopaedia of trees of the world. Oxford University Press, pp. 196–201.
Woldring H (2000) On the origin of plums: a study of sloe, damson, cherry plum, domestic plums and their intermediate forms. Palaeohistoria 39(40):535–562
Zeven AC, De Wet JMJ (1982) Dictionary of cultivated plants and their regions of diversity. Pudoc, Wageningen
Zohary D (1992) Is the European plum, Prunus domestica L., a P. cerasifera Ehrh. x P. spinosa L. allo-polyploid? Euphytica 60:75–77
Acknowledgements
We acknowledge all institutions, repositories and germplasm collections which provided us with plant material for the realisation of this research study. We thank Joseph Rohrer from University of Wisconsin-Eau Claire for his help in determining material from American plums, and Prof. Mario Honrubia and Dr. Almudena Gutierrez from University of Murcia for their support with laboratory equipment. This study was supported by Fundacion Séneca, Centro de Coordinación de la Investigación, Región de Murcia (Spain). Rosaceous genomics is sponsored at East Malling by the BBSRC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Abbott
Rights and permissions
About this article
Cite this article
Reales, A., Sargent, D.J., Tobutt, K.R. et al. Phylogenetics of Eurasian plums, Prunus L. section Prunus (Rosaceae), according to coding and non-coding chloroplast DNA sequences. Tree Genetics & Genomes 6, 37–45 (2010). https://doi.org/10.1007/s11295-009-0226-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-009-0226-9