
Abstract
Poplars (genus Populus) and willows (genus Salix) are members of the Salicaceae, a family of catkin-bearing trees, shrubs and sub-shrubs. Poplar is considered the model system for biological studies in trees and considerable genetic and genomic resources have become available in recent years. The transfer of information to research studies in willow, for which fewer resources are currently available, would be highly beneficial. However, the extent of conservation between poplar and willow genomes has not yet been extensively studied. To address this, we have constructed a linkage map of willow based on a large mapping population derived from a cross between two Salix viminalis × (S. viminalis × S. schwerinii) hybrid sibs, and aligned this to the publicly available poplar genome sequence. A set of genome-wide, expressed poplar sequences was selected and used to design primer sets that efficiently amplified homeologous regions in willow. Direct sequencing of the willow products confirmed homology with the poplar target in the majority of instances and allowed identification of single nucleotide polymorphisms (SNPs) that were used to map these loci. In total, 202 amplified fragment length polymorphisms (AFLPs), 75 microsatellites and 79 SNPs were used to construct a willow consensus map that spanned 1,856.7 cM with an average interval between markers of 6.3 cM. Poplar sequences homologous to those of the mapped willow microsatellite loci were identified and used in addition to the SNP markers to putatively align all but two minor linkage groups to the poplar genome sequence. A high degree of macrosynteny was revealed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Poplars (genus Populus) and willows (genus Salix) are members of the Salicaceae, a family of catkin-bearing trees, shrubs and sub-shrubs. Most are dioecious and have a haploid chromosome number of 19. Populus is considered the model system for genetic and genomic studies in forest trees (Bradshaw et al. 2000; Taylor 2002), largely due to its modest genome size, its fast growth and the ease with which it can be vegetatively propagated. All of these biological traits are shared with Salix. Ongoing interest in poplar has resulted in the development of a considerable range of molecular tools including molecular markers (e.g. van der Schoot et al. 2000; Smulders et al. 2001; Tuskan et al. 2004), genetic maps (e.g. Bradshaw et al. 1994; Wu et al. 2000; Cervera et al. 2001; Yin et al. 2004), expressed sequence tags (ESTs) (Sterky et al. 1998; Wullschleger et al. 2002), microarrays (Hertzberg et al. 2001) and the genome sequence of P. trichocarpa (http://genome.jgi-psf.org/Poptr1/Poptr1.home.html). In comparison, willow is less well studied at the genetic level, although molecular markers (Barker et al. 2003; Hanley 2003) and linkage maps (Tsarouhas et al. 2002; Hanley et al. 2002; Rönnberg-Wästljung et al. 2003; Hanley 2003) have been published in recent years.
Given their close taxonomic relationship, it has been postulated that genetic and genomic information may be readily transferable between Populus and Salix. If true, this would accelerate genetic and complex trait analyses in willow, for which fewer resources are available at present. However, comparative mapping information is currently limited and the true extent of synteny in the Salicaceae is not yet known. The successful transfer of a limited number of molecular markers such as Restriction Fragment Length Polymorphisms (RFLPs) (Tsarouhas et al. 2002) and microsatellites (Hanley et al. 2002; Tuskan et al. 2004) supports the notion that useful levels of nucleotide similarity are conserved between genera. However, the efficiency of successful marker transfer still remains to be fully demonstrated, particularly for microsatellites markers, for which a high degree of sequence conservation in priming sites would be required.
In our experience, the development of microsatellite markers that amplify across Salix is challenging. This is not unexpected, given the high levels of diversity represented in the genus (Barker et al. 1999). Furthermore, depending on the class of underlying repeat motifs, microsatellite loci may be biased towards non-coding regions of the genome (Tóth et al. 2000), which are likely to be less highly conserved between genera than coding sequences (Cato et al. 2001). RFLPs are more tolerant to nucleotide variation and have therefore been the marker system of choice in many comparative studies (e.g. Paterson et al. 2000). However, cross-hybridisation with multiple targets may present difficulties in accurate interpretation of resulting data (Bennetzen 2000). Furthermore, RFLPs are not amenable to high-throughput approaches.
Another problem in comparative studies arises from the fact that multiple sequence homologues may exist in plant genomes, making it difficult to determine whether truly orthologous loci are being compared (Bennetzen 2000). This scenario will likely be encountered in comparisons within the Salicaceae, as analysis of EST data from several poplar species and hybrids indicated that a genome duplication event occurred early in, if not before, the evolution of the Populus genus (Sterck et al. 2005). However, it remains unclear whether this event occurred before the divergence of Populus and Salix. Additional duplication events further back in the ancestry are also likely (Blanc and Wolfe 2004) and evidence for significant genome duplication within Salix has previously been reported (Tsarouhas et al. 2002). Clearly, the occurrence of multiple homologues of several loci within Salicaceae genomes may complicate comparative mapping if markers used are generated without knowledge of the number of copies of their underlying sequence.
The availability of both the P. trichocarpa genomic sequence and a large number of poplar EST sequences has allowed us to address these potential problems and more efficiently develop a set of molecular markers that are both transferable to willow and informative and reliable for comparative study. We selected a genome-wide set of poplar gene sequences that showed little or no homology to any other regions of the poplar genome and amplified and sequenced putative orthologues from the two diploid Salix viminalis × (S. viminalis × S. schwerinii) full-sib parents of a willow mapping population. This large population (n=947), designated K8, (Hanley 2003) is the focus of ongoing linkage mapping and extensive QTL studies (http://www.biomass4energy.org). Polymorphisms identified were then mapped in this pedigree to facilitate a putative alignment of the poplar and willow genomes. In addition, sequences derived from a S. burjatica microsatellite-enriched library (Edwards et al. 1996) previously used to develop microsatellite markers that are now mapped on K8 (Hanley et al. 2002; Barker et al. 2003; Hanley 2003) were compared in silico with the poplar genome to identify additional homologous loci that may be informative in comparison. Details regarding the efficacy of these approaches and resulting genome alignments are presented in this paper, along with further discussion in the context of the implications for future research.
Materials and methods
Plant material
Construction of the willow genetic map was based on the K8 mapping population (Hanley 2003). This family comprises 947 progeny of a cross between two full-sib S. viminalis × (S. viminalis × S. schwerinii) hybrid individuals S3 and R13. Although precise data are difficult to find for all Salix species, based on the limited information available these parents are believed to be of karyotype 2n=38. To limit the amount of screening necessary, a subset of 471 progeny were randomly selected for use in microsatellite and AFLP marker testing and map construction. SNPs were mapped using a randomly selected subset of 96 of these individuals. Genomic DNA was extracted from 50 mg frozen tissue collected from young leaves using the DNeasy96 Plant Extraction Kit (Qiagen) according to the manufacturer’s protocol. DNA quality was assessed by agarose gel electrophoresis and yields were determined by comparison with uncut lambda standards (Sigma) using Quantity One software (BioRad Laboratories).
Microsatellite and AFLP markers
Willow microsatellite markers developed previously (Edwards et al. 1996; Hanley et al. 2002; Barker et al. 2003; Hanley 2003) were first tested for their ability to detect polymorphisms between the K8 parents. A subset of 43 publicly available poplar-derived microsatellite markers that are known to amplify in willow (Tuskan et al. 2004) were also tested. Polymorphic markers were screened against the 471 K8 progeny according to one of two methodologies. Initially, radiolabelling and autoradiography was used as described in Hanley et al. 2002. Later, fluorescent labelling of forward primers was used in the development of PCR and loading multiplexes as described in Hanley 2003. AFLP markers were generated based on the method of Vos et al. (1995) as described in detail in Hanley et al. 2002. Eleven primer combinations were used.
Construction of the willow genetic map
Map construction was performed based on 471 progeny of the K8 mapping population, with JoinMap version 3.0 software (Van Ooijen and Voorrips 2001) used for all aspects of linkage analysis. According to the strategy successfully adopted in Hanley et al. (2002), parental and integrated linkage maps were constructed using a LOD (logarithm of odds) threshold of 4.0 to determine marker groupings, unless otherwise stated. The Kosambi mapping function was used for map construction with the following JoinMap parameter settings: Rec=0.4, LOD=1.0, Jump=5. Resulting linkage maps were drawn using MapChart 2.1 software (Voorrips 2002) and robustness of the consensus groups tested by comparison with the parental maps. To enhance the reliability of the map, the ability of the software to force problematic markers on to the map was not employed. As AFLP markers that are heterozygous in both parents can be unreliable in linkage analysis (Maliepaard et al. 1997; Hanley et al. 2002), markers of this type were excluded from calculations if they led to discrepancies between the parental and consensus map orders, or if they caused difficulty in calculating robust map orders with respect to goodness of fit.
SNP discovery and mapping
Primers were designed to amplify regions of the poplar genome to which poplar EST sequences have been mapped. Only sequences with little or no homology to other regions of the genome were selected. Initially, five roughly equally spaced targets per poplar chromosome were chosen. Chromosome nomenclature used in this paper is according to that employed on the poplar genome website (http://genome.jgi-psf.org/Poptr1/Poptr1.home.html). In all cases, primers were positioned within coding sequence although they were designed to amplify across introns where possible. Primers were not positioned in regions of the reference sequence where there was evidence for polymorphism between the poplar genomic and colocated EST sequences.
The ability to amplify a product from the K8 willow mapping population parents was tested in 20 μl volume PCR reactions containing 10 ng genomic DNA, 2×PCR Master Mix (Promega), 80 ng of both the forward and reverse primer and sterile distilled water to total volume. Thermocycling was performed using a GeneAmp 9700 system (Applied Biosystems) as follows: initial denaturation at 94°C for 1 min, 35 cycles of 94°C for 40 s, 58°C for 1 min, 72°C for 1 min, with a final extension period of 10 min at 72°C. Successful amplification was determined by agarose gel electrophoresis. Where amplification was unsuccessful, or product yields were low, an alternative target in the vicinity of the original was identified and tested.
To determine whether the failure of some primer sets to amplify a product in willow was due to differences in nucleotide sequence between willow and poplar, and not a consequence of unsuccessful primer design, 21 randomly selected primer pairs were also tested on two P. deltoides × P. trichocarpa hybrid sibs. Willow PCR products were extracted from gels using the QIAquick Gel extraction Kit (Qiagen) according to the Manufacturer’s instructions, with the exception that the final elution was performed in sterile distilled water rather than the supplied buffer. Both forward and reverse direction sequencing reactions were performed in 10 μl total volumes comprising two μl BigDye® v1.1 Terminator Reaction Mix (Applied Biosystems), 2 μl 5× BigDye dilution buffer (Applied Biosystems), 100 ng primer, 3 μl PCR product and sterile distilled water to volume. Thermocycling conditions were as follows: 30 cycles of 95°C for 30 s, 50°C for 15 s and 60°C for 3 min. Products were precipitated using the ethanol/sodium acetate protocol supplied by the Manufacturer and resuspended in 10 μl HiDi formamide (Applied Biosystems). Electrophoresis was performed on an ABI PRISM® 3100 Genetic Analyser (Applied Biosystems) according to the Manufacturer’s instructions. Resulting sequences were aligned using the ContigExpress feature of the Vector NTI software package (Invitrogen) and homologies to the original targets were confirmed by BLASTN searches against the poplar genome. Contigs were then examined for the presence of informative polymorphisms for mapping.
Polymorphisms were mapped using a randomly selected subset of 96 K8 progeny using the SNaPshot® Multiplex system (Applied Biosystems). Details of multiplexed SNP assays are provided in Table 1. Each target was individually amplified as previously described although PCR reactions were scaled down to 10 μl total volumes. Equivalent volumes of products amplified with each primer set were then pooled for each individual and 7.5 μl of each resulting pool was incubated with 0.5 U ExoI (New England Biolabs) and 1.25 U shrimp alkaline phosphatase (Amersham Biosciences) at 37°C for 1 h followed by a 15-min inactivation step at 80°C. SNaPshot reactions comprised 2 μl pooled and treated PCR product, 1 μl SNaPshot reaction mix, 0.5 μl pooled primers as described in Table 1, and sterile distilled water to a 5 μl total volume. Thermocycling was performed according to the Manufacturer’s protocol with the exception that 30 cycles were performed. Reactions were then incubated with 1 U calf intestinal phosphatase (Promega) for 1 h at 37°C. One microliter of extension product was mixed with 8.8 μl HiDi formamide and 0.2 μl Genescan™ LIZ-120 size standard (Applied Biosystems) and electrophoresed on an ABI PRISM® 3100 Genetic Analyser according to the Manufacturer’s instructions. Genotypes were determined using GeneMapper® software (Applied Biosystems). All primers used in this study were synthesised and desalted by Sigma-Genosys.
Microsatellite homologies and mapping
Homology searches were performed with sequences of the original S. burjatica-derived microsatellite-enriched library clones (Edwards et al. 1996) against the poplar genome (http://genome.jgi-psf.org/Poptr1/Poptr1.home.html) for all mapped willow-derived microsatellite loci. The BLASTN algorithm (Altschul et al. 1990) was used with an E-value threshold of 0.5 used as a cut-off point. The most significant hit returned for each locus was used to infer a putative position on the poplar genome.
Results
SNP discovery and screening
Of 370 gene-targeted primer sets tested, 169 (45.7%) yielded a single PCR product for both of the willow mapping population parents. Product yields for 26 more primers sets were deemed to be insufficient for direct sequencing, and these loci were excluded from further analysis. Seven (1.9%) primer sets amplified multiple bands and were also discarded. All of the 21 primer sets also tested on poplar amplified a product in both poplar individuals. However, only 11 (52.4%) of these generated a product in willow. Of the 169 loci sequenced, 155 (91.7%) gave high quality sequence reads. The average expected size of these fragments was ∼490 bp. Where sequencing was not successful, amplification of multiple PCR templates from a single primer pair appeared to be the cause. After alignment of the willow-derived sequences for each locus, at least one putative SNP that would be informative for mapping was identified in 103 (66.5%) of the successfully sequenced targets. Homology searches using the polymorphic willow consensus sequences against the poplar genome indicated a most significant hit at the position of the original target in all but three instances. Sequences obtained for V_3 and I_5c showed greatest homology with poplar sequence on chromosome II and scaffold 140, respectively, and not V and I as expected. Willow sequence obtained with primer set V_5b showed homology to poplar chromosome V at a position other than that originally targeted.
Segregation data for 79 SNPs were generated using the SNaPshot multiplex system as described in Table 1. Distorted segregation ratios (P<0.1) were obtained for six SNP markers (Fig. 1). A further five segregating SNP markers that appeared heterozygous in both parents yielded allele ratios that were not consistent with segregation of a single locus (I_4, XIV_2, IV_5, XVI_2b and XVIII_4). In each of these instances, approximately 25% of genotypes appeared homozygous for one allele and 75% were heterozygotes. No homozygotes were detected for the other allele. This could be explained if two homologous loci were amplified in each parent, with each parent being heterozygous at one locus and homozygous for the consistently identified allele at the other. Although it was not possible to determine whether both parents were heterozygous at the equivalent copy, the allele segregating with an approximate 3:1 ratio was scored as a dominant marker and included in the mapping data set. However, as these markers may or may not have represented segregation data from single loci, their inclusion in linkage analysis was treated with caution and the effect of each ‘locus’ examined on an individual basis. Marker V_1 was heterozygous in the female parent and homozygous in the male parent. However, in addition to heterozygous progeny, homozygous progeny were identified for each of the two alleles. The observed segregation ratio was consistent with the presence of a null allele in the male parent. The allele common to both parents was scored as a dominant marker for mapping.


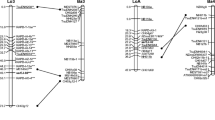
An alignment of the consensus K8 willow linkage map to the poplar genomic sequence based on SNP and microsatellite markers. Markers shown in bold type are those used for alignment. Gene-targeted SNP markers are prefixed with roman numerals, which relate to the poplar chromosome to which they were targeted. Microsatellite markers are prefixed with SB for those developed in willow and ORPM for those derived from poplar. AFLP markers are prefixed with E. Poplar chromosomes are shown on the left of each pair and willow linkage groups on the right, with the approximate positions of markers given in base pairs (bp) and centimorgans (cM), respectively. Loci that may indicate syntenic disparities are underlined. The positions of willow-derived microsatellite markers on poplar chromosomes are based on the most significant BLASTN hit returned. Microsatellite loci in parentheses indicate possible alternative locus positions based on sequence homologies other than the most significant detected. Microsatellites that were assigned to positions that were not in agreement between poplar and willow are indicated in italics. Two willow linkage groups (A and B) that could not be aligned to the poplar genome are also shown. The predicted positions of six unlinked SNP markers are also shown. Markers showing segregation distortion are indicated by asterisks on the willow map (one asterisk: P<0.1, two asterisks: P<0.05, three asterisks: P<0.01, four asterisks: P<0.005, five asterisks: P<0.001, six asterisks: P<0.0005, seven asterisks: P<0.0001)
Construction of the willow maps
In total, 204 AFLP markers were included in the mapping data set. Of these, 57 were derived from the female parent, 52 were from the male parent and 95 were heterozygous in both parents. Of 72 polymorphic willow-derived microsatellites also included, 20 segregated in the female parent only, 20 segregated in the male parent only, 21 were heterozygous for the same two alleles in both parents and 11 were heterozygous for a different allele in each respective parent and were therefore fully informative. Successful amplification was achieved for 37 of the 43 poplar-derived microsatellites tested. Of these, markers ORPM030, ORPM327 and ORPM446 gave scorable polymorphic profiles in the willow family and were also included in the mapping data. Of the 79 segregating SNPs, 29 were maternally informative, 24 were paternally informative and 26 segregated in both parents.
Construction of the maternal map was based on 57 AFLP markers, 29 SNPs and 33 microsatellites. Marker groupings using a LOD threshold of 4.0 resulted in the identification of 17 linkage groups containing three or more markers, four duplets and eight unlinked markers. Calculation of marker orders resulted in a maternal linkage map that spanned 843.2 cM with an average interval between markers of 9.3 cM. For the paternal map, groupings based on 52 AFLP, 32 microsatellites and 24 SNPs resulted in 13 linkage groups, with six duplets and eight markers remaining unlinked. Two highly distorted (P<0.0001) AFLP markers, EataMaat02 and EagcMaag13, were subsequently removed from the paternal data set before final groupings as no linked markers showed evidence of distortion and thus they may have comprised superimposed loci that co-migrated to equivalent positions on the gels. Calculated linkage groups spanned 812.0 cM with an average interval between markers of 10.3 cM.
In total, 358 segregating markers were included for construction of the consensus map. The data set comprised 202 AFLP markers, 75 microsatellites and 79 SNP markers. Marker groupings at a LOD threshold of 4.0 identified 24 linkage groups comprising three or more markers, seven duplets and 11 markers remained unlinked. Re-calculation of linkage groups after exclusion of the SNPs that were scored as dominant markers did not lead to changes in the observed groupings. Therefore, these markers were retained in subsequent analysis. As more than 19 linkage groups were obtained, marker grouping were also performed using a LOD of threshold of 3.0 in an attempt to improve resolution. However, this resulted in the identification of 14 groups comprising three or more markers, suggesting that spurious linkages were obtained. However, microsatellite SB1279, which was unlinked to any other marker at LOD 4.0, joined group XVI without suggestion of spurious linkage. This grouping was therefore used in subsequent map calculation. Consensus linkage groups could not be calculated for two groupings (XV and XIX) due to a paucity of data from markers segregating in both parents. These two groups were split into the parental groups and the markers segregating in both parents tested separately with each parental group in subsequent marker order calculations.
Six of the gene-targeted SNP markers remained unlinked to any other markers in the data set (IV_1c, VII_5c, VIII_1d, XIII_5d, XVI_5b and X_4). The remaining 75 gene-targeted SNP markers grouped together with only markers derived from the equivalent poplar linkage group in all but five instances. Markers XIV_3d and XIV_5d, originally designed to target poplar linkage group XIV, showed linkage with SNPs derived from poplar chromosome II, despite significant homologies being detected only on chromosome XIV in both cases. In agreement with its most significant homology, marker V_3 also appeared linked to group II. Similarly, marker I_5c, which showed greatest homology to unmapped scaffold 140, grouped with markers targeted to chromosome XI. Marker XI_5 showed linkage with markers derived from poplar chromosome I. This was not concurrent with its expected grouping (on XI) based on sequence homology.
After calculation of marker orders for each consensus linkage group, resulting orders were in agreement with those calculated for the parental maps with one exception. The position of the paternal AFLP marker EagaMaag04 differed between the paternal and consensus linkage groups. This marker had several missing genotypes and was therefore discarded from analysis. An additional 14 AFLP markers that segregated in both parents were removed from map calculations as they caused problems with respect to goodness of fit. The resulting linkage groups of the consensus map are shown in Fig. 1 and spanned 1,856.7 cM with an average interval between markers of 6.3 cM.
Microsatellite homologies
Results of BLASTN homology searches for the 72 mapped willow microsatellite loci are shown in Table 2. All but two loci (SB918 and SB1045) showed significant homology with the poplar genomic sequence data. Based on the most significant homology returned for each locus, a likely position on the poplar genome could be inferred for 55 loci. Putative homeologous positions in poplar could not be determined for 15 additional microsatellites that showed greatest homology to unmapped poplar genomic scaffolds that are yet to be assigned to a specific genomic location.
The relative assignment of microsatellites to linkage groups was in agreement between the willow map generated here and the poplar genomic sequence with the following exceptions. Locus SB274, which mapped to willow linkage group II, showed greatest homology to sequence on poplar chromosome XIV. Loci SB988, SB984, SB276, SB80 all SB1035 all showed greater homology to poplar linkage groups that were not homeologous to the willow linkage groups to which they mapped in this study. However, for the latter three loci, hits returned were based on short sequence homologies and thus BLASTN scores were low in these instances. The sequence of locus SB988 showed greatest homology to poplar chromosome XIX, although a second highly significant hit (E value: 1e-064), which was in agreement with its relative willow map position on linkage group VI, was also obtained.
Marker SB984 showed significant homology to poplar chromosome III exclusively. This marker formed a duplet with VII_3b, which only showed significant homology to the sequence from which it was derived on poplar chromosome VII. The sequence of willow microsatellite locus SB514b showed greatest homology to poplar chromosome II. However, no evidence for linkage of this marker to others assigned to the homeologous willow group was detected. The linkage group to which SB514b mapped in willow (group A) comprised only AFLP markers and could not be aligned to the poplar genome. However, a second significant homology (E value: 2.0e-48) for this microsatellite sequence was detected on poplar chromosome IV in a region to which no willow linkage groups were aligned in this study (Fig. 1).
Alignment of the willow map to the poplar genome
The alignment of consensus willow linkage groups to the poplar genome sequence is shown in Fig. 1. Of the 31 willow linkage groups and duplets identified, all but two were putatively aligned to the poplar genome. The orientation of four putatively aligned willow groups anchored by a single SNP or microsatellite marker could not be determined relative to poplar (i.e. VIa, VIIb, VIIIb and XIIIa). Based on the available SNP marker data, synteny between genomes was generally maintained with the exception of a single region. Here, markers XIV_3d and XIV_5d, which were originally targeted to poplar chromosome XIV, mapped to willow linkage group II. This syntenic disparity was supported by microsatellite locus SB274, which also mapped to willow linkage group II despite a most significant homology on poplar chromosome XIV (Table 2). The possibility of spurious linkage was eliminated by testing markers XIV_3d, XIV5d and SB274 for linkage with only those markers mapping to willow group XIV. No evidence for linkage was detected at a LOD threshold of 3.0, suggesting that the observed groupings with markers on willow group II are likely to be robust. Marker XIV_3d was excluded from final map order calculations, as it could not be placed without utilising the ability of the software to force it on to the linkage group.
After accounting for the final positions of markers V_3 and I_5c, for which homology results suggested that primers amplified targets other than those intended, the only SNP marker that was non-syntenic was XI_5. This marker mapped to willow linkage group I at a position that may have been represented by the third most significant BLASTN result (Fig. 1).
Discussion
The alignment of the K8 willow map to the poplar genomic sequence represents the first comparative mapping study within the Salicaceae. The K8 mapping population was used to underpin this study, as its large size affords greater potential map resolution and robustness of linkages compared with the smaller populations used in previous willow linkage mapping efforts (n=92, Rönnberg-Wästljung et al. 2003; n=66, Hanley et al. 2002; n=87, Tsarouhas et al. 2002). In this study, 471 individuals were used to generate segregation data for AFLP and microsatellite markers. These marker classes were used for construction of the K8 linkage map for reasons outlined in Hanley et al. (2002).
The consensus map was used for alignment with the poplar sequence, as the genome coverage was far greater than that achieved for each of the parental maps. Although information provided by dominant markers heterozygous in both parents may be limited in mapping studies involving outbreeders (Maliepaard et al. 1997), this class of marker was included in analysis to improve genome coverage, as a large proportion of these markers would have been expected in the K8 progeny because it was generated from a cross between full-sibs. Furthermore, their inclusion aided calculation of consensus linkage groups, in turn, increasing the resolution afforded by the alignment. However, to ensure robustness of the K8 map in the future, it may be prudent to replace these markers with more informative alternatives as they become available.
The alignment revealed macrosyntenic relationships between genomes with all but two of the willow linkage groups being putatively assigned to homologous poplar chromosomes. However, additional markers will be required to confirm alignments of willow linkage groups that contained only a single comparative marker (i.e. VIa, VIIc, VIIIb and XIIIa), particularly in instances where the marker used as a basis for alignment was a microsatellite. Similarly, the true map positions of unlinked SNP markers, although inferred in this study, will require confirmation. The correct alignment of VIIb also remains to be established. This duplet comprised a SNP marker and a microsatellite, which gave conflicting comparative mapping results. The SNP marker was used in preference of the microsatellite for alignment in this study, as this marker type is more likely to be reliable. However, this assignment may need to be revised as additional data become available.
The assignment of multiple willow linkage groups to single poplar chromosomes was a common feature of the data. This allowed further resolution of the willow map, as unlinked groups that may originate from an equivalent chromosome could be identified. No evidence of linkage was detected between these willow linkage groups at the LOD threshold employed for two possible reasons. First, a paucity of markers in regions between linkage groups corresponding to an equivalent chromosome will have prevented detection of linkage. In some instances, these gaps may represent regions of the genome with high rates of recombination. For example, willow linkage groups Ia and Ib were both assigned to poplar chromosome I but were not linked using a LOD threshold of 4.0, despite both linkage groups being fairly well represented by markers. It is, therefore, possible that the interval between markers EagaMacc15 and EagcMaat26 is subject to elevated levels of recombination and additional marker saturation within this region will be required to confirm linkage. Markers mapping to these two groups were tested for linkage using a LOD threshold of 3.0. Although linkage was detected, insufficient segregation data were available to calculate a combined group. Second, several willow linkage groups (e.g. those corresponding to poplar chromosomes VI, VII and XVII) comprised only markers derived from a single parent. In the absence of markers heterozygous in both parents within these regions, linkage between these groups would not have been detected. As highlighted by willow linkage groups XV and XIX, which were linked at a LOD threshold of 4.0 but had to be split into parental groupings for reliable marker ordering, further resolution of the K8 map should prove possible, as additional markers become available.
The reason why several of the SNP markers failed to show linkage to any of the willow linkage groups became clear after alignment. With the exception of one marker, all were targeted to regions of the poplar genome that were not represented by the willow map. In addition, the ability to detect linkage for the SNP markers will have been compromised in comparison to the AFLPs and microsatellites, as they were only screened against a subset of the mapping progeny.
The observation that all but three gene-targeted markers mapped to positions that were in possible agreement with their original targets suggested that the approach used in their design successfully delivered reliable markers for comparative study. However, as a minority of these primers amplified a sequence other than that intended, care should still be taken to confirm sequence homology when transferring sequence information between genera. Furthermore, as exemplified by marker XI_5, mapping should be used to support claims of orthology.
Results obtained for markers SB274, XIV_3d and XIV_5d, which all mapped to willow linkage group II but showed greatest nucleotide homology to poplar chromosome XIV, may indicate a disparity between the willow and poplar genomes. Further investigation will be required to determine the reason for this result, as several explanations are feasible based on the currently available data. However, this observation highlights the possibility that the assignment of some willow linkage groups to poplar chromosomes, particularly those that comprised relatively few comparative markers, may have been based on paralogues that exist in duplicated genomic regions. This could easily occur if a copy of a target is deleted in either genome. For this reason, assignments and nomenclature adopted for some willow linkage groups in this study may require revision as further data become available.
The use of poplar coding sequence for the development of primers that amplified in willow was relatively efficient, with more than half of primer pairs successfully generating a product in willow. The observation that the majority of primer sets generated a single band in willow was consistent with the selection of only those poplar sequences with little or no nucleotide homology to any other region currently represented in the genome sequence data. Direct sequencing of the willow PCR products provided support that putative orthologues were amplified and also provided an efficient and reliable route to SNP discovery. The identification of at least one SNP that was informative for mapping in approximately two thirds of sequenced loci is commensurate with the outbreeding/heterozygous nature of the Salix genus (Barker et al. 2003). In addition, the SNP polymorphism detection rate (66.5%) was greater than that achieved with microsatellite markers in previous willow mapping studies on populations K3 (Hanley et al. 2002) and K8 (Hanley 2003) and with the poplar microsatellite primers tested here, of which only ∼10% of markers that amplified in willow were informative in the K8 population. This finding is in agreement with the fact that SNPs are the most abundant class of polymorphism in most genomes (Kwok et al. 1996; Kruglyak 1997).
The SNP markers generated in this study potentially afforded coverage of much of the poplar genome with four exceptions. Poplar chromosomes IV, V, VIII and XII had large regions that were not represented. Data generated in this study suggest that homeologous segments are likely to exist in the willow genome as three, three, four and three loci derived from these areas, respectively, were successfully amplified and sequenced in willow. These sequences showed homology to their intended targets but could not be mapped, as polymorphisms were not detected.
The efficiency of direct transfer of microsatellite markers between poplar and willow for comparative mapping has previously been examined. Hanley et al. (2002) tested 19 poplar derived primer sets but found only five (26%) of these generated a PCR product in willow. More recently, Tuskan et al. (2004) reported that between 30 and 50% of poplar microsatellite primer sets developed in their study amplified a product in willow, with success being dependent on the species tested. The observation in this present study that 86% of poplar-derived primer sets tested generated a PCR product in willow is not comparable with these previous estimates, as loci were only tested in this study if they were already known to work in some willow species. The fact that six of these primer sets did not amplify in the K8 parents may indicate that species in this pedigree (S. viminalis and S. schwerinii) are more distantly related to poplar than those included in the study of Tuskan et al. (2004) and therefore, direct transfer of poplar markers to this pedigree may be less efficient.
The use of willow microsatellite library sequence homology for comparative mapping largely overcame the limitations of primer transfer between genera, as the requirement for conservation in priming sites and the reliance on polymorphism was bypassed. Although the value of this approach was supported by the large proportion of mapped loci (∼76%) for which the most significant homology returned was concurrent with their willow map position, a greater number of discrepancies were observed using microsatellites in comparison to the gene-targeted SNPs, with seven of the 70 microsatellite sequences that returned significant hits showing greatest homology to regions of the poplar genome that did not correspond to their respective willow map positions. Mapping of the genomic scaffolds in poplar will shed further light on the efficiency of this method in the future. However, in both willow and poplar, a single microsatellite primer pair may amplify different regions of the genome in different mapping populations and species (Cervera et al. 2001), or may amplify multiple loci in a single mapping population (Hanley 2003; Yin et al. 2004). Therefore, the occurrence of multiple priming sites for some microsatellites in Salicaceae genomes may complicate interpretation of comparative mapping results. Furthermore, for comparisons based on sequence homology alone, the possibility that paralogues may be compared in some instances should not be dismissed.
The availability of multiple comparative markers on the majority of willow linkage groups permitted a preliminary examination of colinearity, although resolution was clearly limited by the number and position of markers available. However, as micro-colinearity between orthologous regions of the Arabidopsis and Populus genomes has been reported (Stirling et al. 2003), it may be reasonable to predict that this is upheld at a greater level between Salix and Populus genomes, which are more closely related in terms of phylogeny (Sterck et al. 2005). In general, colinearity was largely maintained, although there were some exceptions. In most cases, disparities were between closely linked markers (e.g. SB1303 and SB1048) and may be the result of mapping errors, as suggested in other comparative mapping studies (e.g. Chagné et al. 2003). However, as the population size used in the current study afforded a high degree of mapping power, the possibility that these are true breakdowns in colinearity should not yet be discounted. The mapping of additional markers or the use of genotype information from additional K8 progeny may shed light on the validity of these results in the future.
Consequences of this study
The results of this study highlight a number of potentially important consequences for genome analyses within the Salicaceae. We have demonstrated that there is sufficient conservation of coding nucleotide sequence between genera to permit efficient development of markers for comparative studies. While not all poplar-derived primer sets yielded a product in willow in this study, our experience suggests that modifying the position of the primers within the target sequence can often lead to successful amplification (unpublished data). It should therefore be possible to isolate functional candidate gene sequences from willow using the public poplar genome and EST resources as a basis. However, problems regarding amplification of paralogous sequences instead of true orthologues must be considered and mapping should be used to support suggestions of orthology. Furthermore, the levels of synteny and colinearity observed in this study imply that it will often be possible to develop targeted markers for mapping in willow based on corresponding positions in poplar. This will vastly improve the efficiency with which gaps can be filled and provide an efficient route to increasing marker density in regions of interest. Furthermore, the resulting alignment will permit comparative trait mapping between genera and may provide a route towards identification of genes underlying QTL through positional candidate gene approaches.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Barker JHA, Matthes M, Arnold GM, Edwards KJ, Åhman I, Larsson S, Karp A (1999) Characterisation of genetic diversity in potential biomass willows (Salix spp.) by RAPD and AFLP analyses. Genome 42:173–183
Barker JHA, Pahlich A, Trybush S, Edwards KJ, Karp A (2003) Microsatellite markers for diverse Salix species. Mol Ecol Notes 3:4–6
Bennetzen JL (2000) Comparative sequence analysis of plant nuclear genomes: microcolinearity and its many exceptions. Plant Cell 12:1021–1029
Blanc G, Wolfe KH (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16:1667–1678
Bradshaw HD Jr, Villar M, Watson BD, Otto KG, Stewart S, Stettler RF (1994) Molecular genetics of growth and development in Populus. III. A genetic linkage map of hybrid poplar composed of RFLP, STS and RAPD markers. Theor Appl Genet 89:167–178
Bradshaw HD, Ceulemans R, Davis J, Stettler R (2000) Emerging model systems in plant biology: Poplar (Populus) as a model forest tree. J Plant Growth Regul 19:306–313
Cato SA, Gardner RC, Kent J, Richardson TE (2001) A rapid PCR-based method for genetically mapping ESTs. Theor Appl Genet 102:296–306
Cervera MT, Storme V, Ivens B, Gusmão J, Liu BH, Hostyn V, Van Slycken M, Van Montague M, Boerjan W (2001) Dense genetic linkage maps of three Populus species (Populus deltoides, P. nigra and P. trichocarpa) based on AFLP and microsatellite markers. Genetics 158:787–809
Chagné D, Brown G, Lalanne C, Madur D, Pot S, Neale D, Plomion C (2003) Comparative genome and QTL mapping between maritime and loblolly pines. Mol Breed 12:185–195
Edwards KJ, Barker JHA, Daly A, Jones C, Karp A (1996) Microsatellite libraries enriched for several microsatellite sequences in plants. Biotechniques 20:758–760
Hanley SJ (2003) Genetic mapping of important agronomic traits in biomass willow. Ph.D. thesis, University of Bristol, UK
Hanley S, Barker JHA, Van Ooijen JW, Aldam C, Harris SL, Åhman I, Larsson S, Karp A (2002) A genetic linkage map of willow (Salix viminalis) based on AFLP and microsatellite markers. Theor Appl Genet 105:1087–1096
Hertzberg M, Aspeborg H, Schrader J, Andersson A, Erlansson R, Blomqvist K, Bhalerao R, Uhlén M, Teeri TT, Lundeberg J, Sundberg B, Nilsson P, Sandberg G (2001) A transcriptional roadmap to wood formation. Proc Natl Acad Sci U S A 98:14732–14737
Kruglyak L (1997) The use of a genetic map of biallelic markers in linkage studies. Nat Genet 138:499–510
Kwok PY, Deng Q, Zakeri H, Taylor SL, Nickerson DA (1996) Increasing the information content of STS-based genome maps: identifying polymorphisms in mapped STSs. Genomics 31:123–126
Maliepaard C, Jansen J, Van Ooijen JW (1997) Linkage analysis in a full-sib family of an outbreeding species: overview and consequences for applications. Genet Res 70:237–250
Paterson AH, Bowers JE, Burow MD, Draye X, Elsik CG, Jiang C-X, Katsar CS, Lan T-H, Lin Y-R, Ming R, Wright RJ (2000) Comparative genomics of plant chromosomes. Plant Cell 12:1523–1539
Rönnberg-Wästljung AC, Tsarouhas V, Semerikov V, Lagercrantz U (2003) A genetic linkage map of a tetraploid Salix viminalis × S. dasyclados hybrid based on AFLP markers. For Genet 10:185–194
Smulders MJM, van der Schoot J, Arens P, Vosman B (2001) Trinucleotide repeat microsatellite markers for black poplar (Populus nigra L.). Mol Ecol Notes 1:188–190
Sterck L, Rombauts S, Jansson S, Sterky F, Rouzé P, Van de Peer Y (2005) EST data suggest that poplar is an ancient polyploid. New Phytol 167:165–170
Sterky F, Regan S, Karlsson J, Hertzberg M, Rohde A, Holmberg A, Amini B, Bhalerao R, Larsson M, Villarroel R, Van Montegu M, Sandberg G, Ollson O, Teeri TT, Boerjan W, Gustafsson P, Uhlén M, Sundberg B, Lundeberg J (1998) Gene discovery in the wood-forming tissues of poplar: analysis of 5,692 expressed sequence tags. P Natl Acad Sci U S A 95:13330–13335
Stirling B, Koo Yang Z, Gunter LE, Tuskan GA, Bradshaw HD Jr (2003) Comparative sequence analysis between orthologous regions of the Arabidopsis and Populus genomes reveals substantial synteny and microcollinearity. Can J For Res 33:2245–2251
Taylor G (2002) Populus: Arabidopsis for forestry. Do we need a model tree? Ann Bot 90:681–689
Tóth G, Gáspári Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10:967–981
Tsarouhas V, Gullberg U, Lagercrantz (2002) An AFLP and RFLP linkage map and quantitative trait locus (QTL) analysis of growth traits in Salix. Theor Appl Genet 105:277–288
Tuskan GA, Gunter LE, Yang ZK, Yin TM, Sewell MM, DiFazio SP (2004) Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa. Can J For Res 34:85–93
van der Schoot J, Popiskova M, Vosman B, Smulders MJM (2000) Development and characterization of microsatellite markers in black poplar (Populus nigra L.). Theor Appl Genet 101:317–322
Van Ooijen JW, Voorrips RE (2001) JoinMap® 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Voorrips RE (2002) MapChart: software for the graphical representation of linkage maps and QTLs. J Hered 93:77–78
Vos P, Hogers R, Bleeker M, Reijans M, Vandelee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP—a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wu RL, Han YF, Hu JJ, Fang KK, Li L, Li ML, Zeng ZB (2000) An integrated genetic map of Populus deltoides based on amplified fragment length polymorphisms. Theor Appl Genet 100:1249–1256
Wullschleger SD, Jansson S, Taylor G (2002) Genomics and forest trees: Populus emerges as the perennial favourite. Plant Cell 14:2651–2655
Yin TM, DiFazio SP, Gunter LE, Riemenschneider S, Tuskan GA (2004) Large-scale heterospecific segregation distortion in Populus revealed by a dense genetic map. Theor Appl Genet 109:451–463
Acknowledgements
This study is part of the BEGIN (Biomass for Energy Genetic Improvement Network) project (bioass4energy.org) funded by the Department of Environment, Food and Rural Affairs (DEFRA) (Project no. NF0424) and the Biotechnology and Biological Sciences Research Council of the United Kingdom. We would like to thank Prof Gail Taylor, University of Southampton, UK for helpful discussions and for providing the poplar leaf material used in this study. Rothamsted Research receives grant-aided support from the Biotechnology and Biological Sciences Research Council of the United Kingdom.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hanley, S.J., Mallott, M.D. & Karp, A. Alignment of a Salix linkage map to the Populus genomic sequence reveals macrosynteny between willow and poplar genomes. Tree Genetics & Genomes 3, 35–48 (2006). https://doi.org/10.1007/s11295-006-0049-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-006-0049-x