
Abstract
This review examines the recent models describing the mode of action of various xylanolytic enzymes and how these enzymes can be applied (sequentially or simultaneously) with their distinctive roles in mind to achieve efficient xylan degradation. With respect to homeosynergy, synergism appears to be as a result of β-xylanase and/or oligosaccharide reducing-end β-xylanase liberating xylo-oligomers (XOS) that are preferred substrates of the processive β-xylosidase. With regards to hetero-synergism, two cross relationships appear to exist and seem to be the reason for synergism between the enzymes during xylan degradation. These cross relations are the debranching enzymes such as α-glucuronidase or side-chain cleaving enzymes such as carbohydrate esterases (CE) removing decorations that would have hindered back-bone-cleaving enzymes, while backbone-cleaving-enzymes liberate XOS that are preferred substrates of the debranching and side-chain-cleaving enzymes. This interaction is demonstrated by high yields in co-production of xylan substituents such as arabinose, glucuronic acid and ferulic acid, and XOS. Finally, lytic polysaccharide monooxygenases (LPMO) have also been implicated in boosting whole lignocellulosic biomass or insoluble xylan degradation by glycoside hydrolases (GH) by possibly disrupting entangled xylan residues. Since it has been observed that the same enzyme (same Enzyme Commission, EC, classification) from different GH or CE and/or AA families can display different synergistic interactions with other enzymes due to different substrate specificities and properties, in this review, we propose an approach of enzyme selection (and mode of application thereof) during xylan degradation, as this can improve the economic viability of the degradation of xylan for producing precursors of value added products.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass is made up of three major components, namely, cellulose (40–60%), hemicellulose (20–40%) and lignin (10–25%) (Kang et al. 2014). Hemicelluloses are the second most abundant terrestrial polysaccharide in nature, after cellulose, and represent about 20–40% of the dry weight of the lignocellulosic biomass. Hemicellulose is a hetero-polysaccharide composed of pentoses (xylose and arabinose), hexoses (mannose, glucose and galactose) and sugar acids (methyl-glucuronic acid) (Saha 2003). Hemicelluloses are usually grouped into classes based on the main sugar residues present in the backbone, and these include xyloglucans, xylans, mannans and glucomannans, and β-glucans (Scheller and Ulvskov 2010).
Over the past few decades, xylan has attracted much attention for its industrial importance in the bioconversion of lignocellulosic biomass to biofuels, pulp and paper, food, improvement of animal feedstock digestibility, production of pre-biotic oligosaccharides and organic synthesis of value added products (VAPs) such as xylitol (Guo et al. 2013; Kamat et al. 2013; Samanta et al. 2015; Charoensiddhi et al. 2017). Enzymatic hydrolysis of plant carbohydrates such as xylan using xylanolytic enzymes has emerged as the most prominent technology for the conversion of biomass into VAPs (Van Dyk and Pletschke 2012).
Xylanolytic enzymes are produced by a variety of micro-organisms including bacteria, fungi and yeast, which are naturally found in diverse niches such as insect guts, manure, waste water, hot environmental samples and chicken cecum to name a few (Kumar et al. 2016; Chadha et al. 2019). In addition to the production of a variety of xylanolytic enzymes, many micro-organisms produce multiple isoforms of the same class of xylanolytic enzyme with different properties (Collins et al. 2005; Malgas et al. 2017; Chadha et al. 2019). It is noteworthy to mention that not all micro-organisms produce all the enzymes required in a xylanolytic arsenal for efficient xylan degradation. For instance, Thermomyces lanuginosus only produces a single GH11 xylanase, while Thermomyces stellatus produces ten xylanases from GH families 10, 11 and 30 (Chadha et al. 2019). With a huge reservoir of xylanolytic enzymes available, it is important to know which enzymes would be required (i.e. enzymes with high stability and activity, cooperability with other enzymes and tolerance to inhibitors) for efficient xylan degradation in industry for high yields of VAP production.
Recent studies have demonstrated synergistic interactions between various xylanolytic enzymes that occur and improve xylan and xylan-containing biomass bioconversion into the precursor sugars required for VAP synthesis. Here, we summarize recent studies and hypotheses on how the glycoside hydrolase (GH) families, auxiliary activity enzymes (LPMOs and carbohydrate esterases) and mode of application of the xylanolytic enzymes affect their synergistic associations during xylan degradation for industrial applications.
Xylan structure
The structure of xylan is quite variable, ranging from linear backbones constituted of β-1,4-linked poly-xylose residues denoted as homo-xylans to branched hetero-xylans, whereby the prefix ‘hetero’ denotes the presence of branching sugar residues other than D-xylose. The branching sugars in hetero-xylans consist of either L-arabinofuranose linked to the C-3 positions of D-xylose residues, these hetero-xylans are denoted as arabinoxylans (AX) (Schendel et al. 2015), or of D-glucuronic acid or its methyl derivative, 4-O-methyl-D-glucuronic acid, linked to the C-2 positions of D-xylose residues, these hetero-xylans are denoted as glucuronoxylans (GX) (Togashi et al. 2009), and/or a mixture of both sugars, these hetero-xylans denoted as arabinoglucuronoxylans (AGX) (Ebringerová 2006). The α-L-arabinofuranosyl units in hetero-xylans may further be substituted by hydroxycinnamates (i.e. ferulic acid and p-coumaric acid) at the C-5 position (Schendel et al. 2015). The degree and type of branching sugar residues in hetero-xylans vary depending on the plant source of the xylan in question. Xylans of several hardwoods are acetylated at either the C-2 or C-3 position and/or at both of these positions of D-xylose residues (Pinto et al. 2005). Xylan and lignin are generally considered to be linked through covalent bonds to form lignin–carbohydrate complexes (LCCs). In the case of GX, cross linkages are formed between 4-O-methyl-D-glucuronic acid residues and aliphatic chains of lignin (Li and Helm 1995).
Xylan degradation
A consortium of xylanolytic Carbohydrate-Active enzymes (CAZymes), including glycoside hydrolases (GH) and auxiliary activity enzymes [i.e. carbohydrate esterases (CE) and lytic polysaccharide monooxygenases (LPMOs)], are required for complete hetero-xylan degradation in lignocellulosic biomasses. In this section, we provide a summary of the biochemical properties of the enzymes required for hetero-xylan degradation.
Xylanases (EC 3.2.1.8) are key enzymes for xylan degradation, they act on the xylan backbone, cleaving randomly at internal sites on β-1,4-glycosidic linkages between xylopyranosyl residues. Since xylanases randomly cleave the xylan backbone, their action products are diverse and may include xylobiose, xylotriose, xylotetraose, longer and/or branched xylo-oligomers (XOS). Xylanases belong to several GH families i.e. family 5, 8, 9, 10, 11, 16, 30, 43, 51 and 98 in the CAZy database (www.cazy.org). Some GH5 subfamily 34 arabinoxylanases have been reported to possess a decoration requirement which shows catalytic specificity for arabinose-substituted xylans, generating XOS branched at the reducing end (Karlsson et al. 2018). GH8 xylanases are mechanistically distinct from other xylanase GH families (retaining mechanism) as they are the only family which executes catalysis via a net inversion mechanism. GH10 xylanases are highly active on short XOS, soluble and branched xylans (Biely et al. 2016; Karlsson et al. 2018). GH11 xylanases, on the other hand, are highly active on unsubstituted regions of xylan and insoluble xylan, with diminished activity on decorated xylans (Mendis and Simsek 2015). GH30 subfamily 8 xylanases have been shown to be dependent on methyl-glucuronic acid substitutions for activity, and as a result, are called glucuronoxylanases (Biely et al. 2014, 2015).
Recently discovered oligosaccharide reducing-end xylanases, also called Rexs (EC. 3.2.1.156), are strictly found in family GH8 (Valenzuela et al. 2016). Rexs hydrolyse the xylan backbone or XOS from the reducing ends producing short XOS and xylose (Lagaert et al. 2007; Juturu and Wu 2012; Malgas and Pletschke 2019). The xylosidases (E.C 3.2.1.37), on the other hand, hydrolyse the xylanase and Rex-produced XOS by continuously removing D-xylose residues from the non-reducing termini, but do not hydrolyse xylan (Yan et al. 2008; Knob et al. 2010; Huy et al. 2015). Xylosidases are presently found in families GH 1, 2, 3, 5 (Subfamily 22), 30, 39, 43, 51, 52, 54, 116 and 120.
Alpha-arabinofuranosidases, also called Abfs (EC 3.2.1.55), hydrolyse substituents of α-L-arabinofuranoside (L-Araf) residues in AXs and arabinosyl substituted XOS (Lagaert et al. 2014). Abfs belong to GH families 2, 3, 10, 43, 51, 54 and 62. Abfs that release L-Araf units from mono-substituted main-chain D-xylopyranosyl (D-Xylp) motifs are termed AXH-m, while those that release single L-Araf residues from double-substituted main-chain D-Xylp motifs are termed AXH-d. Only GH43 and 51 Abfs have been demonstrated to readily de-branch L-Araf on both mono- and di-substituted D-Xylp motifs (Lagaert et al. 2014). Alpha-glucuronidases (EC 3.2.1.139), on the other hand, hydrolyse α-(1,2)-D-(4-O-methyl)-glucuronosyl side chains in the main chains of GXs and AGXs. Generally, GH67 glucuronidases prefer short glucuronic acid substituted XOS and are generally intracellular or membrane associated (Nagy et al. 2002; Nurizzo et al. 2002; Golan et al. 2004), while GH115 glucuronidases are more active on polymeric glucuronic acid-containing xylans, these being consequently extracellularly produced enzymes (Tenkanen and Siika-Aho 2000; McKee et al. 2016).
LCCs pose a barrier to xylan hydrolysis by glycosyl hydrolases and lead to a major limitation on the utilisation of plant biomass as a renewable source (Jeffries 1990). Therefore, efficient enzymatic hydrolysis of xylan also requires the carbohydrate esterases (CEs) such as feruloyl esterases (FAEs), glucuronoyl esterases (GEs) and acetyl xylan esterases (AXEs). Acetyl xylan esterases (EC 3.1.1.72) are mainly found in CE families 1–7 and 16 in the CAZy database and are primarily involved in the hydrolysis of ester bonds to liberate acetic acid from acetylated polysaccharides, making the main chain accessible to glycoside hydrolase enzymes (Zhang et al. 2011). Feruloyl esterases (E.C. 3.1.1.73), on the other hand, are classified into CE family 1 (Lei et al. 2016). FAEs catalyse the cleavage of ester bonds between a hydroxycinnamate and AX, releasing a phenolic acid such ferulic acid or p-coumaric acid (Wong et al. 2013). Finally, glucuronoyl esterases (EC3.1.1.-) belong to CE family 15; GEs cleave ester bonds between lignin aliphatic alcohols and the 4-O-methyl-D-glucuronic acid substituents of GX (Baath et al. 2016).
Recently, lytic polysaccharide monooxygenases (LPMOs) or Auxiliary activity proteins (AAs, EC 1.14.99.53-54/56) have been discovered and hold major potential for future biomass degradation applications (Horn et al. 2012; Arfi et al. 2014). These enzymes enhance glycoside hydrolase activity by cleaving glycosidic bonds of polysaccharides in the crystalline and insoluble portion via an oxidative mechanism, consequently resulting in complete polysaccharide degradation (Cragg et al. 2015; Müller et al. 2015). LPMOs are currently sequence-classified into four families in the CAZy database. Members of the AA9 family are mainly active on cellulose with a few reported to be active on hemicellulose. On the other hand, members from family AA10 exhibit activity on cellulose or chitin. Members from AA11 and AA13 families are active on chitin and starch, respectively.
A number of comprehensive reviews on the biochemical characteristics of xylanolytic enzymes (Knob et al. 2010; Juturu and Wu 2012; Lagaert et al. 2014) and non-GH activity CAZymes (Hemsworth et al. 2015; Oliveira et al. 2019) have been published over the past decade, this review will focus on recent advances in understanding the synergism which occurs between xylanolytic enzymes during the degradation of xylans in lignocellulosic biomass.
Synergistic actions of xylanolytic enzymes during xylan hydrolysis
As mentioned before, a consortium of xylan specific CAZymes synergistically act on xylans, leading to their complete hydrolysis into monomeric sugars, acids and mono-phenols. Synergism between hemicellulolytic enzymes is characterised into two modes, namely homeosynergy (that between two backbone-cleaving enzymes or two side-chain-cleaving xylanolytic enzymes) and heterosynergy (that between a side-chain-cleaving and a main-chain-cleaving enzyme) (Moreira and Filho 2008). Based on the complexity of xylan, which includes the presence of substituents on its backbone, xylanolytic enzymes are expected to exhibit these two types of synergisms during its hydrolysis.
Homeosynergy studies between glycoside hydrolases during xylan hydrolysis
Synergism between xylanolytic enzymes such as xylanases, exo-xylanases and xylosidases is vital for efficient xylan backbone degradation and the production of xylose (see Table 1). Synergistic combinations of (1) xylanase with xylanase and (2) exo-xylanase (Rex) with xylanase(s) have been investigated and generally lead to higher production of XOS. As mentioned previously, xylanases of different GH families show varied substrate specificities (Christakopoulos et al. 2003; Beaugrand et al. 2004; Broeker et al. 2018), therefore, combining different GH family xylanases generally leads to a diversity and higher production of XOS (Goncalves et al. 2015; Malgas and Pletschke 2019). On the other hand, the Rex with xylanase synergism is as a result of the Rex showing a different specificity compared to xylanases; (1) acting from the reducing end of xylans and (2) on the xylanase generated XOS to produce smaller XOS and xylose (Liu et al. 2019a; Malgas and Pletschke 2019). The xylosidases then further hydrolyse the XOS that are produced by both Rex and xylanases into xylose (see Table 1). Interestingly, xylan backbone degradation is not a linear process, but a relational loop between the enzymes involved as Liu and co-workers showed higher synergism between xylanase and xylosidase when the enzymes are applied simultaneously compared to when applied sequentially (Liu et al. 2019b). We now know that XOS inhibit glycoside hydrolases and hamper biomass degradation (Kumar and Wyman 2009; Qing et al. 2010). Therefore, hydrolysis of XOS by xylosidases, which are their preferred substrates compared to xylans, should reduce product inhibition of xylanases—leading to a complementary relationship between the two enzyme classes whereby xylosidases further hydrolyse the XOS that are produced by both exo- and endo-xylanases (after product inhibition alleviation) into xylose.
Heterosynergy studies between glycoside hydrolases during xylan hydrolysis
Generally, with regards to heterosynergistic interactions between backbone- and side-chain-cleaving enzymes, two cross relationships appear to lead to synergism between these enzymes during hetero-xylan degradation (see Table 2). Firstly, synergism appears to result when a side-chain-cleaving enzyme (i.e. GH115 glucuronidase) removes decorations on hetero-xylans that would have hindered the action of a backbone cleaving enzyme (i.e. xylanases) from liberating XOS (Yang et al. 2015). It is noteworthy to mention that, to date, there is no Abf reported to display activity on intact xylan polymers, therefore, the above-mentioned cross relationship may be only applicable to xylanases and GH115 glucuronidases. This type of synergism leads to a higher production of XOS when these enzymes are applied, and has been reported to work both simultaneously and sequentially (see Table 2). Secondly, synergism also occurs when a main-chain-cleaving enzyme liberates decorated XOS from hetero-xylans (which are preferred substrates compared to polymeric xylan) for side-chain-cleaving enzymes (Rosa et al. 2013). This type of synergism leads to a higher production of the hetero-xylan decorating sugars such as L-Araf and 4-O-methyl-glucuronic acid. Interestingly, combining GH67 glucuronidases with GH10 xylanases always yields synergism, however, combining them with GH11 xylanases doesn’t lead to synergism (Rosa et al. 2013; Rhee et al. 2017) (see Table 2). It is likely that the GH10 xylanase produces 4-O-methyl-glucuronic acid terminal substituted XOS that are a more preferred substrate for GH67 glucuronidase than the internally substituted XOS produced by GH11 xylanases (Karlsson et al. 2018). However, the GH115 derived glucuronidases appear to tolerate the GH11 xylanase-produced 4-O-methyl-glucuronic acid substituted XOS, as synergism is observed between these enzymes (Rhee et al. 2017).
Synergy studies on xylan hydrolysis by GHs and axillary activity CAZymes
A few studies have been reported in literature where glycoside hydrolases have been used in conjunction with axillary activity CAZymes (i.e. CEs or LPMOs) for improved hetero-xylan degradation. Here we give a summary of the findings from these studies.
CEs exhibit a strong synergistic relationship with xylanases for degradation of various lignocellulosic substrates (see Table 2). It appears that the removal of side groups by CEs from the xylan main chain and on branching sugars on the xylans increases accessibility of xylanases to the substrate and improves the production of XOS. Hydrolysis of xylan by xylanases is often restricted in the presence of acetyl substituents. AXEs are known for their ability to remove acetyl substituents from the xylan main chain and thereby increase accessibility of xylanases to xylan. A number of studies have reported on synergism between AXEs and xylanases for hydrolysis of different substrates (Yang and Liu 2008; Zheng et al. 2013). Secondly, CEs (mainly FAEs) seem to prefer short substituted XOS generated by xylanase as substrates for cleaving ester bonds releasing FA. Currently, there is still very limited information on the synergistic action of GEs and xylanases during degradation of plant biomass as only one study has been reported to date (Mosbech et al. 2018).
Recent studies have broadened the known substrate specificity of LPMOs to include isolated xylan and XOS—such findings have been reported for AA9-family LPMOs from Lentinus similis (LsAA9A) and Collariella virescens (CvAA9A) (Simmons et al. 2017), and Myceliophthora thermophila C1 (MtLPMO9A) (Frommhagen et al. 2015). The implications of these findings are the potential application of LPMOs synergistically with glycoside hydrolases for the degradation of xylans and xylan containing lignocellulosic biomass. Indeed, LPMOs have been shown to synergise with xylanases during the degradation of isolated xylans (Kim et al. 2016; Sanhueza et al. 2018) and xylan-containing lignocellulosic biomass (Jung et al. 2015; Kim et al. 2016). Recently, for the first time, an actinomycete, Kitasatospora papulose, AA10-family LPMO has been shown to boost xylose release by a xylanase in beechwood xylan and by Celluclast (a xylanase containing enzyme preparation) on sugarcane bagasse (Corrêa et al. 2019). The synergism between these two enzyme classes is suggested to be due to the mode of action of LPMOs which leads to loosening of the rigid xylan–cellulose polysaccharide matrix in plant biomass and a reduction in inter-microfibril bonding, enabling increased accessibility to the matrix for glycoside hydrolases (Frommhagen et al. 2015; Hu et al. 2018).
A proposed up-to-date model of xylan degradation
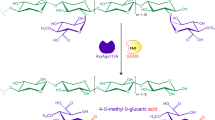
A proposed up-to-date general scheme showing how xylanolytic enzymes mechanistically degrade hetero-xylans (i.e. AGXs) in a synergistic fashion is presented below (Fig. 1). Firstly, GH11 xylanases preferably cleave unsubstituted regions of the xylan backbone or insoluble xylans (Kumar et al. 2016). GH11 xylanases may be sterically hindered by the presence of acetyl groups on the xylopyranosides constituting the xylan backbone; this then necessitates the action of CE1 acetyl xylan esterases to remove these groups on the xylan backbone to allow xylanase action to proceed (Adesioye et al. 2016). Secondly, GH10 xylanases (Xyn) cleave highly decorated AGX backbones or the soluble XOS produced by GH11 Xyn and release XOS which contain arabinose or glucuronic side chains at the non-reducing end (Karlsson et al. 2018). On the other hand, GH115 glucuronidases (Agu) are efficient at removing the methyl-glucuronic acid residues from non-terminal positions of both XOS and polymeric xylans (Rhee et al. 2017). The GH115 Agu action removes substituents which otherwise would have sterically hindered GH10/11 Xyn activity during xylan degradation. The GH8 Rex can then hydrolyse xylan from the reducing ends and on the GH10/11 Xyn-produced XOS, leading to the production of shorter XOS and xylose (Malgas and Pletschke 2019). However, arabinofuranosidases and XOS specific glucuronidases may be required for debranching the GH10-produced XOS before Rex can act on them. The arabinofuranosidases (AXH-d) release single L-Araf residues from double-substituted XOS while AXH-m release single L-Araf residues from single-substituted XOS, on the other hand, GH67 glucuronidases (Agu67A) prefer short methyl-glucuronic acid substituted XOS (Lagaert et al. 2014). Regions rich with arabinose and/or glucuronic acid substituents may require the action of both GH5 arabinoxylanase and GH30 glucuronoxylanase, respectively, to cleave these xylans into highly substituted XOS (Biely et al. 2016; Karlsson et al. 2018). These branched XOS can then be acted upon by the debranching enzymes, AXH and Agu67A, respectively. Finally, GH43 xylosidases (Xyl) release xylose from the non-reducing ends of XOS, and as a result, alleviate product inhibition to xylanases by their products (XOS).

An illustration of the enzyme sites of the enzymes required to completely degrade a hetero-xylans and b hetero-xylans associated to cellulose fibers. Snip diagonal corner rectangles represent xylose, pentagons represent arabinose, hexagons represent (methyl)-glucuronic acid residues, triangles represent acetyl groups, circles represent hydroxycinnamate groups (i.e. ferulic acid or p-coumaric acid), stars represent aliphatic alcohol groups and cylinders represent cellulose chains. The arrows represent glycosidic (thick arrows) or ester bonds (thin arrows) recognized by the respective enzymes and the rectangle represents regions of xylan which are flat and in a twofold helical screw ribbon bound to cellulose microfibrils
A recent review by Biely and co-workers postulated that the addition of side-chain-cleaving enzymes such as α-glucuronidase to a mixture of xylanase and xylosidase (and even Rex) would bring about only small changes in the saccharification yield of xylan (Biely et al. 2016). The reason for this is that some xylan fragments remain esterified with acetic acid. The authors, therefore, postulated that, on de-glucuronylated oligosaccharides, the action of xylosidase would stop at the first acetylated Xylp residue from the non-reducing end as a result, enzymatic saccharification cannot proceed to completion without deacetylation of XOS by acetyl xylan esterases (AXEs) (Biely et al. 2016). CE4/7/16 AXEs have been reported to specifically de-acetylate XOS (Adesioye et al. 2016, 2018). The role of CE16 may be particularly critical for the efficient degradation of the mono-acetylated aldotetraouronic acid (3′′-Ac3MeGlcA3Xyl3), generated from acetyl-GX degradation by GH10 Xyn, as it is one of the most enzyme resistant acetyl-GX fragments to Agu and Xyl action (Puchart et al. 2016). More CEs, specifically targeting esterified compounds (i.e. hydroxycinnamate groups or lignin) on side chain sugars found on xylan such as arabinose and glucuronic acid, may also be required as debranching enzymes are restricted by these esterified compounds.
In addition to CEs, it has recently been shown that LPMOs are critical in boosting xylan degradation into monosaccharides by GHs (Jung et al. 2015; Kim et al. 2015; Corrêa et al. 2019). Several studies have reported that in a given xylan, the linear and unsubstituted fractions tend to self-associate so as to become aggregated into insoluble aggregates that can interact with crystalline cellulose (Linder et al. 2003; Hu et al. 2018). Interestingly, a recent study has shown that xylans are susceptible to AA14 oxidative cleavage only when they are adsorbed onto crystalline cellulose, and not when they are in solution (Couturier et al. 2018). This has been attributed to recalcitrant xylans bound to cellulose microfibrils displaying a two-fold screw axis conformation aligned parallel to the cellulose chain direction, which seems to be compatible with the proper orientation of the carbohydrate H1 and H4 atoms with respect to the LPMO catalytic center (Linder et al. 2003; Couturier et al. 2018). These xylan-xylan and xylan-cellulose entanglement junctions are believed to be inaccessible to glycoside hydrolases, therefore, the action of LPMOs is crucial in reducing these inter-microfibril bonding sites to allow accessibility of xylanolytic glycoside hydrolases to act on the xylan polysaccharides found in these regions. As it is known that LPMOs are copper dependent enzymes requiring an electron donor, recently, the mechanism and origin of the electron supply for these enzymes in biological systems such as plant biomass has been shown to be from long-range electron transfer involving soluble low molecular weight lignins produced by ligninases (i.e. laccases and peroxidases) from high molecular weight lignins (Westereng et al. 2015). The implication of these findings is that xylan degradation may, to some degree, be linked to lignin modification, particularly with respect to electron donation for LPMO activity efficiency.
It is clear that with the entire consortium of xylanolytic enzymes, including accessory/non-GH enzymes such as esterases and LPMOs, hetero-xylan hydrolysis with monomeric sugar yields as high as 100% can be attained. We have compiled a list of all the enzymes which, to date, have been shown to be essential for the efficient degradation of AGX (see Table 3).The aforementioned model of xylan hydrolysis by xylanolytic enzymes will hopefully shed insights into the selection of not only the necessary enzyme classes required, but also the specific families as described in the CAZy database and the sequence (sequential versus simultaneous) of application of these enzymes in an industrial setting in order to achieve high yields of VAP production from xylans and xylan-containing biomass. This will hopefully lead to a significant improvement in the economic viability of the bioconversion of xylan—containing lignocellulosic biomass into various VAPs. It is noteworthy to mention that industrial application of these xylanolytic enzyme cocktails demands that the enzymes must be able to withstand harsh processing conditions such as acidic/alkaline conditions, elevated temperatures, and the presence of biomass pre-treatment by-products and enzyme reaction products which could be inhibitory to these enzymes in the catalysis process (Malgas et al. 2017; Chadha et al. 2019). Therefore, caution regarding the aforementioned conditions should be taken during the selection of xylanolytic enzymes to constitute an enzyme cocktail for lignocellulosic biomass degradation.
Conclusion and future perspectives
In this review, it was demonstrated that the complex structure of xylans presents a great challenge to degradation. A model consisting of multiple xylanolytic enzyme activities which are required to fully hydrolyse xylans was then proposed. It was clearly shown that hydrolysis of xylans to completion is most likely obtained through the synergistic interactions of complementary endo-, exo- and side-chain-acting-xylanolytic activities complemented by auxiliary activity enzymes (CEs and LPMOs). We are hopeful that the proposed model of xylan degradation by xylanolytic enzymes will inform industry of a suitable approach for the application of these enzymes in an industrial setting in order to achieve high yields of VAP production from xylans and xylan-containing biomass. However, limitations and knowledge gaps in the field of xylan degradation by enzymatic means are also highlighted in this review. Listed below are but a few:
-
1.
Metagenomic mining for novel CAZymes with unique properties regarding xylan degradation is still not fully explored.
-
2.
The characterisation of xylanolytic enzymes—both biochemically and according to families in the CAZy database—requires more work on enzymes such GH5 arabinoxylanases, GH8 Rex or exo-xylanases, GH30 glucuronoxylanases, CE15 GEs and LPMOs (AA9-10, 14), as their catalytic properties and mechanisms are still unclear.
-
3.
It may be of interest to probe into the implications of lignin modifying enzymes (i.e. laccases and peroxidases) for their influence on xylan degradation.
-
4.
Some of the remaining challenges in the field of biomass degradation include the use of protein engineering to produce new enzyme functionalities such as broad substrate specificity, tolerance to products and pre-treatment by-product inhibition, and thermal stability.
-
5.
Finally, the design of chimeric multi-functional enzymes and enzyme complexes (i.e. artificial xylanosomes) has not been well explored compared to that of the design of cellulosomes for cellulose degradation. Such work could greatly advance xylan degradation.
Exploration of such research avenues can lead to an improvement in the efficiency of xylan degradation and this would subsequently lead to an improvement in the economic viability of the bioconversion of high xylan-containing lignocellulosic biomass into fermentable sugars for bioethanol and the production of other fine chemicals.
Abbreviations
- AA:
-
Auxiliary activity
- Abf:
-
α-Arabinofuranosidase
- Agu:
-
α-Glucuronidase
- AGX:
-
Arabinoglucuronoxylan
- AX:
-
Arabinoxylan
- AXE:
-
Acetyl xylan esterase
- AXH-d:
-
Doubly substituted L-Araf specific α-arabinofuranosidase
- AXH-m:
-
Mono-substituted L-Araf specific α-arabinofuranosidase
- CAZy:
-
Carbohydrate active enzyme database
- CAZyme:
-
Carbohydrate-active enzyme
- CE:
-
Carbohydrate esterase
- DS:
-
Degree of synergy
- EC:
-
Enzyme commission number
- FA:
-
Ferulic acid
- FAE:
-
Feruloyl esterase
- GE:
-
Glucuronoyl esterase
- GH:
-
Glycoside hydrolase
- GX:
-
Glucuronoxylan
- LPMO:
-
Lytic polysaccharide mono-oxygenase
- Rex:
-
Oligosaccharide reducing-end xylanase
- RS:
-
Reducing sugar(s)
- XOS:
-
Xylo-oligosaccharide(s)
- Xyn:
-
β-Xylanase
- Xyl:
-
β-Xylosidase
References
Adesioye FA, Makhalanyane TP, Biely P, Cowan DA (2016) Phylogeny, classification and metagenomic bioprospecting of microbial acetyl xylan esterases. Enzyme Microb Technol 93–94:79–91
Adesioye FA, Makhalanyane TP, Vikram S et al (2018) Structural characterization and directed evolution of a novel acetyl xylan esterase reveals thermostability determinants of the carbohydrate esterase 7 family. Appl Environ Microbiol 84(8):e02695–e2717
Arfi Y, Shamshoum M, Rogachev I et al (2014) Integration of bacterial lytic polysaccharide monooxygenases into designer cellulosomes promotes enhanced cellulose degradation. Proc Natl Acad Sci 111:9109–9114
Baath J, Giummarella N, Klaubauf S et al (2016) A glucuronoyl esterase from Acremonium alcalophilum cleaves native lignin-carbohydrate ester bonds. FEBS Lett 590:2611–2618
Banka AL, Albayrak Guralp S, Gulari E (2014) Secretory expression and characterization of two hemicellulases, xylanase, and beta-xylosidase, isolated from Bacillus subtilis M015. Appl Biochem Biotechnol 174:2702–2710
Beaugrand J, Chambat G, Wong VWK et al (2004) Impact and efficiency of GH10 and GH11 thermostable endoxylanases on wheat bran and alkali-extractable arabinoxylans. Carbohydr Res 339:2529–2540
Biely P, MalovÓková A, Hirsch J et al (2015) The role of the glucuronoxylan carboxyl groups in the action of endoxylanases of three glycoside hydrolase families: a study with two substrate mutants. Biochim Biophys Acta - Gen Subj 1850:2246–2255
Biely P, Puchart V, Stringer MA, Mørkeberg Krogh KBR (2014) Trichoderma reesei XYN VI - A novel appendage-dependent eukaryotic glucuronoxylan hydrolase. FEBS J 281:3894–3903
Biely P, Singh S, Puchart V (2016) Towards enzymatic breakdown of complex plant xylan structures: state of the art. Biotechnol Adv 34(7):1260–1274
Broeker J, Mechelke M, Baudrexl M et al (2018) The hemicellulose-degrading enzyme system of the thermophilic bacterium Clostridium stercorarium: comparative characterisation and addition of new hemicellulolytic glycoside hydrolases. Biotechnol Biofuels 11(1):229
Chadha BS, Kaur B, Basotra N et al (2019) Thermostable xylanases from thermophilic fungi and bacteria: current perspective. Bioresour Technol 277:195–203
Charoensiddhi S, Conlon MA, Franco CMM, Zhang W (2017) The development of seaweed-derived bioactive compounds for use as prebiotics and nutraceuticals using enzyme technologies. Trends Food Sci Technol 70:20–33
Choengpanya K, Arthornthurasuk S, Wattana-Amorn P et al (2015) Cloning, expression and characterization of β-xylosidase from Aspergillus Niger ASKU28. Protein Expr Purif 115:132–140
Christakopoulos P, Katapodis P, Kalogeris E et al (2003) Antimicrobial activity of acidic xylo-oligosaccharides produced by family 10 and 11 endoxylanases. Int J Biol Macromol 31:171–175
Cobucci-Ponzano B, Strazzulli A, Iacono R et al (2015) Novel thermophilic hemicellulases for the conversion of lignocellulose for second generation biorefineries. Enzyme Microb Technol 78:63–73
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29:3–23
Corrêa LTR, Júnior AT, Wolf LD et al (2019) An actinobacteria lytic polysaccharide monooxygenase acts on both cellulose and xylan to boost biomass saccharification. Biotechnol Biofuels 12(1):117
Couturier M, Ladevèze S, Sulzenbacher G et al (2018) Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat Chem Biol 14:306–310
Cragg SM, Beckham GT, Bruce NC et al (2015) Lignocellulose degradation mechanisms across the tree of life. Curr Opin Chem Biol 29:108–119
Ebringerová A (2006) Structural diversity and application potential of hemicelluloses. Macromol Symp 232:1–12
Faundez C, Perez R, Ravanal MC, Eyzaguirre J (2019) Penicillium purpurogenum produces a novel, acidic, GH3 beta-xylosidase: heterologous expression and characterization of the enzyme. Carbohydr Res 482:107728
Frommhagen M, Sforza S, Westphal AH et al (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol Biofuels 8:101
Ghio S, Ontañon O, Piccinni FE et al (2018) Paenibacillus sp. A59 GH10 and GH11 extracellular endoxylanases: application in biomass bioconversion. BioEnergy Res 11:174–190
Golan G, Shallom D, Teplitsky A et al (2004) Crystal structures of Geobacillus stearothermophilus alpha-glucuronidase complexed with its substrate and products: mechanistic implications. J Biol Chem 279:3014–3024
Goncalves GAL, Takasugi Y, Jia L et al (2015) Synergistic effect and application of xylanases as accessory enzymes to enhance the hydrolysis of pretreated bagasse. Enzyme Microb Technol 72:16–24
Guerfali M, Gargouri A, Belghith H (2011) Catalytic properties of Talaromyces thermophilus alpha-L- arabinofuranosidase and its synergistic action with immobilized endo-beta-1,4-xylanase. J Mol Catal B 68:192–199
Guo X, Zhang R, Li Z et al (2013) A novel pathway construction in Candida tropicalis for direct xylitol conversion from corncob xylan. Bioresour Technol 128:547–552
Hemsworth GR, Johnston EM, Davies GJ, Walton PH (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol 33(12):747–761
Hettiarachchi SA, Kwon YK, Lee Y et al (2019) Characterization of an acetyl xylan esterase from the marine bacterium Ochrovirga pacifica and its synergism with xylanase on beechwood xylan. Microb Cell Fact 18:122
Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG (2012) Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45
Hu J, Tian D, Renneckar S, Saddler JN (2018) Enzyme mediated nanofibrillation of cellulose by the synergistic actions of an endoglucanase, lytic polysaccharide monooxygenase (LPMO) and xylanase. Sci Rep 8:3195
Huy ND, Le NC, Seo JW et al (2015) Putative endoglucanase PcGH5 from Phanerochaete chrysosporium is a beta-xylosidase that cleaves xylans in synergistic action with endo-xylanase. J Biosci Bioeng 119:416–420
Jamaldheen SB, Thakur A, Moholkar VS, Goyal A (2019) Enzymatic hydrolysis of hemicellulose from pretreated Finger millet (Eleusine coracana) straw by recombinant endo-1,4-beta-xylanase and exo-1,4-beta-xylosidase. Int J Biol Macromol 135:1098–1106
Jeffries TW (1990) Biodegradation of lignin-carbohydrate complexes. Biodegradation 1:163–176
Jia X, Mi S, Wang J et al (2014) Insight into glycoside hydrolases for debranched xylan degradation from extremely thermophilic bacterium Caldicellulosiruptor lactoaceticus. PLoS ONE 9:1–12
Jung S, Song Y, Myeong H, Bae H (2015) Enhanced lignocellulosic biomass hydrolysis by oxidative lytic polysaccharide monooxygenases (LPMOs) GH61 from Gloeophyllum trabeum. Enzyme Microb Technol 77:38–45
Juturu V, Teh TM, Wu JC (2014) Expression of Aeromonas punctata ME-1 exo-xylanase X in E. coli for efficient hydrolysis of xylan to xylose. Appl Biochem Biotechnol 174:2653–2662
Juturu V, Wu JC (2012) Microbial xylanases: engineering, production and industrial applications. Biotechnol Adv 30:1219–1227
Kamat S, Khot M, Zinjarde S et al (2013) Coupled production of single cell oil as biodiesel feedstock, xylitol and xylanase from sugarcane bagasse in a biorefinery concept using fungi from the tropical mangrove wetlands. Bioresour Technol 135:246–253
Kambourova M, Mandeva R, Fiume I et al (2007) Hydrolysis of xylan at high temperature by co-action of the xylanase from Anoxybacillus flavithermus BC and the beta-xylosidase/alpha-arabinosidase from Sulfolobus solfataricus Oα. J Appl Microbiol 102:1586–1593
Kang Q, Appels L, Tan T, Dewil R (2014) Bioethanol from Lignocellulosic Biomass: Current Findings Determine Research Priorities. Sci World J. https://doi.org/10.1155/2014/298153
Karlsson EN, Schmitz E, Linares-pastén JA, Adlercreutz P (2018) Endo-xylanases as tools for production of substituted xylooligosaccharides with prebiotic properties. Appl Microbiol Biotechnol 102:9081–9088
Kim IJ, Nam KH, Yun EJ et al (2015) Optimization of synergism of a recombinant auxiliary activity 9 from Chaetomium globosum with cellulase in cellulose hydrolysis. Appl Microbiol Biotechnol 99:8537–8547
Kim IJ, Youn HJ, Kim KH (2016) Synergism of an auxiliary activity 9 (AA9) from Chaetomium globosum with xylanase on the hydrolysis of xylan and lignocellulose. Process Biochem 51:1445–1451
Knob A, Terrasan CRF, Carmona EC (2010) β-Xylosidases from filamentous fungi: an overview. World J Microbiol Biotechnol 26:389–407
Kormelink FJM, Voragen AGJ (1993) Degradation of different [(glucurono)arabino]xylans by a combination of purified xylan-degrading enzymes. Appl Microbiol Biotechnol 38:688–695
Kumar R, Wyman C (2009) Effects of cellulase and xylanase enzymes on the deconstruction of solids from pretreatment of poplar by leading technologies. Biotechnol Prog 25:302–314
Kumar V, Marín-Navarro J, Shukla P (2016) Thermostable microbial xylanases for pulp and paper industries: trends, applications and further perspectives. World J Microbiol Biotechnol 32:1–10
Lagaert S, Pollet A, Courtin CM, Volckaert G (2014) β-Xylosidases and α-L-arabinofuranosidases: accessory enzymes for arabinoxylan degradation. Biotechnol Adv 32:316–332
Lagaert S, Van Campenhout S, Pollet A et al (2007) Recombinant expression and characterization of a reducing-end xylose-releasing exo-oligoxylanase from Bifidobacterium adolescentis. Appl Environ Microbiol 73:5374–5377
Lei Z, Shao Y, Yin X et al (2016) Combination of xylanase and debranching enzymes specific to wheat arabinoxylan improve the growth performance and gut health of broilers. J Agric Food Chem 64:4932–4942
Li K, Helm RF (1995) Synthesis and rearrangement reactions of ester-linked lignin-carbohydrate model compounds. J Agric Food Chem 43:2098–2103
Linder Å, Bergman R, Bodin A, Gatenholm P (2003) Mechanism of assembly of xylan onto cellulose surfaces. Langmuir 19:5072–5077
Liu X, Jiang Z, Liu Y et al (2019a) Biochemical characterization of a novel exo-oligoxylanase from Paenibacillus barengoltzii suitable for monosaccharification from corncobs. Biotechnol Biofuels 12:190
Liu Y, Huang L, Zheng D et al (2019b) Biochemical characterization of a novel GH43 family beta-xylosidase from Bacillus pumilus. Food Chem 295:653–661
Long L, Zhao H, Ding D et al (2018) Heterologous expression of two Aspergillus niger feruloyl esterases in Trichoderma reesei for the production of ferulic acid from wheat. Bioprocess Biosyst Eng 41:593–601
Makela MR, Dilokpimol A, Koskela SM et al (2018) Characterization of a feruloyl esterase from Aspergillus terreus facilitates the division of fungal enzymes from carbohydrate esterase family 1 of the carbohydrate-active enzymes (CAZy) database. Microb Biotechnol 11:869–880
Malgas S, Pletschke BI (2019) The effect of an oligosaccharide reducing-end xylanase, Bh Rex8A, on the synergistic degradation of xylan backbones by an optimised xylanolytic enzyme cocktail. Enzyme Microb Technol 122:74–81
Malgas S, Thoresen M, van Dyk SJ, Pletschke BI (2017) Time dependence of enzyme synergism during the degradation of model and natural lignocellulosic substrates. Enzyme Microb Technol 103:1–11
Martins MP, Ventorim RZ, Coura RR et al (2018) The beta-xylosidase from Ceratocystis fimbriata RM35 improves the saccharification of sugarcane bagasse. Biocatal Agric Biotechnol 13:291–298
McKee LS, Sunner H, Anasontzis GE et al (2016) A GH115 α-glucuronidase from Schizophyllum commune contributes to the synergistic enzymatic deconstruction of softwood glucuronoarabinoxylan. Biotechnol Biofuels 9:2
Mendis M, Simsek S (2015) Production of structurally diverse wheat arabinoxylan hydrolyzates using combinations of xylanase and arabinofuranosidase. Carbohydr Polym 132:452–459
Moreira LRS, Filho EXF (2008) An overview of mannan structure and mannan-degrading enzyme systems. Appl Microbiol Biotechnol 79:165–178
Mosbech C, Holck J, Meyer AS, Agger JW (2018) The natural catalytic function of CuGE glucuronoyl esterase in hydrolysis of genuine lignin – carbohydrate complexes from birch. Biotechnol Biofuels 11:71
Müller G, Várnai A, Johansen KS et al (2015) Harnessing the potential of LPMO-containing cellulase cocktails poses new demands on processing conditions. Biotechnol Biofuels 8:187
Nagy T, Emami K, Fontes CMG et al (2002) The membrane-bound α-glucuronidase from Pseudomonas cellulosa hydrolyzes 4-O-methyl-D-glucuronoxylooligosaccharides but not 4-O- methyl-D-glucuronoxylan. J Biotechnol 184:4925–4929
Nurizzo D, Nagy T, Gilbert HJ, Davies GJ (2002) The structural basis for catalysis and specificity of the Pseudomonas cellulosa alpha-glucuronidase, GlcA67A. Structure 10:547–556
Oliveira DM, Mota TR, Oliva B et al (2019) Feruloyl esterases : biocatalysts to overcome biomass recalcitrance and for the production of bioactive compounds. Bioresour Technol 278:408–423
Pinto PC, Evtuguin DV, Neto CP (2005) Structure of hardwood glucuronoxylans: modifications and impact on pulp retention during wood kraft pulping. Carbohydr Polym 60:489–497
Puchart V, Agger JW, Berrin J-G et al (2016) Comparison of fungal carbohydrate esterases of family CE16 on artificial and natural subtrates. J Biotechnol 233:228–236
Qing Q, Yang B, Wyman CE (2010) Xylooligomers are strong inhibitors of cellulose hydrolysis by enzymes. Bioresour Technol 101:9624–9630
Raweesri P, Riangrungrojana P, Pinphanichakarn P (2008) α-L-Arabinofuranosidase from Streptomyces sp. PC22: purification, characterization and its synergistic action with xylanolytic enzymes in the degradation of xylan and agricultural residues. Bioresour Technol 99:8981–8986
Rhee MS, Sawhney N, Kim YS et al (2017) GH115 α-glucuronidase and GH11 xylanase from Paenibacillus sp. JDR-2: potential roles in processing glucuronoxylans. Appl Microbiol Biotechnol 101:1465–1476
Rogowski A, Baslé A, Farinas CS et al (2014) Evidence that GH115 α-glucuronidase activity, which is required to degrade plant biomass, is dependent on conformational flexibility. J Biol Chem 289:53–64
Romano de Carvalho D, Carli S, Meleiro LP et al (2018) A halotolerant bifunctional beta-xylosidase/alpha-L-arabinofuranosidase from Colletotrichum graminicola: purification and biochemical characterization. Int J Biol Macromol 114:74–750
Rosa L, Ravanal MC, Mardones W, Eyzaguirre J (2013) Characterization of a recombinant α-glucuronidase from Aspergillus fumigatus. Fungal Biol 117:380–387
Saha BC (2003) Hemicellulose bioconversion. J Ind Microbiol Biotechnol 30:279–291
Samanta AK, Jayapal N, Jayaram C et al (2015) Xylooligosaccharides as prebiotics from agricultural by-products : production and applications. Bioact Carbohydrates Diet Fibre 5:62–71
Sanhueza C, Carvajal G, Soto-aguilar J et al (2018) The effect of a lytic polysaccharide monooxygenase and a xylanase from Gloeophyllum trabeum on the enzymatic hydrolysis of lignocellulosic residues using a commercial cellulase. Enzyme Microb Technol 113:75–82
Santos CR, Hoffmam ZB, Peixoto V et al (2014) Molecular mechanisms associated with xylan degradation by Xanthomonas plant pathogens. J Biol Chem 289:32186–32200
Scheller HV, Ulvskov P (2010) Hemicelluloses. Annu Rev Plant Biol 61:263–289
Schendel RR, Becker A, Tyl CE, Bunzel M (2015) Isolation and characterization of feruloylated arabinoxylan oligosaccharides from the perennial cereal grain intermediate wheat grass (Thinopyrum intermedium). Carbohydr Res 407:16–25
Sheng P, Xu J, Saccone G et al (2014) Discovery and characterization of endo-xylanase and beta-xylosidase from a highly xylanolytic bacterium in the hindgut of Holotrichia parallela larvae. J Mol Catal B 105:33–40
Shi P, Chen X, Meng K et al (2013) Distinct actions by Paenibacillus sp. strain E18 α-larabinofuranosidases and xylanase in xylan degradation. Appl Environ Microbiol 79:1990–1995
Simmons TJ, Frandsen KEH, Ciano L et al (2017) Structural and electronic determinants of lytic polysaccharide monooxygenase reactivity on polysaccharide substrates. Nat Commun 8:1064
Tenkanen M, Siika-Aho M (2000) An α-glucuronidase of Schizophyllum commune acting on polymeric xylan. J Biotechnol 78:149–161
Togashi H, Kato A, Shimizu K (2009) Enzymatically derived aldouronic acids from Eucalyptus globulus glucuronoxylan. Carbohydr Polym 78:247–252
Valenzuela SV, Lopez S, Biely P et al (2016) The glycoside hydrolase family 8 reducing-end xylose-releasing exo-oligoxylanase Rex8A from Paenibacillus barcinonensis BP-23 is active on branched xylooligosaccharides. Appl Environ Microbiol 82:5116–5124
Van Dyk JS, Pletschke BI (2012) A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes-Factors affecting enzymes, conversion and synergy. Biotechnol Adv 30:1458–1480
Wang L, Shi H, Xu B et al (2016) Characterization of Thermotoga thermarum DSM 5069 alpha-glucuronidase and synergistic degradation of xylan. BioResources 11:5767–5779
Wefers D, Cavalcante JJV, Schendel RR et al (2017) Biochemical and structural analyses of two cryptic esterases in Bacteroides intestinalis and their synergistic activities with cognate xylanases. J Mol Biol 429:2509–2527
Westereng B, Cannella D, Agger JW et al (2015) Enzymatic cellulose oxidation is linked to lignin by long-range electron transfer. Sci Rep 5:18561
Wong DWS, Chan VJ, Liao H, Zidwick MJ (2013) Cloning of a novel feruloyl esterase gene from rumen microbial metagenome and enzyme characterization in synergism with endoxylanases. J Ind Microbiol Biotechnol 40:287–295
Yan QJ, Wang L, Jiang ZQ et al (2008) A xylose-tolerant β-xylosidase from Paecilomyces thermophila: characterization and its co-action with the endogenous xylanase. Bioresour Technol 99:5402–5410
Yang C, Liu W (2008) Purification and properties of an acetylxylan esterase from Thermobifida fusca. Enzyme Microb Technol 42:181–186
Yang W, Bai Y, Yang P, et al. (2015) A novel bifunctional GH51 exo-α-L-arabinofuranosidase/endo-xylanase from Alicyclobacillus sp. A4 with significant biomass-degrading capacity. Biotechnol Biofuels 8: 197.
Yang X, Shi P, Huang H et al (2014a) Two xylose-tolerant GH43 bifunctional β-xylosidase/α-arabinosidases and one GH11 xylanase from Humicola insolens and their synergy in the degradation of xylan. Food Chem 148:381–387
Yang X, Shi P, Ma R et al (2014b) A new GH43 alpha-arabinofuranosidase from Humicola insolens Y1: biochemical characterization and synergistic action with a xylanase on xylan degradation. Appl Biochem Biotechnol 175:1960–1970
Zhang J, Siika-Aho M, Tenkanen M, Viikari L (2011) The role of acetyl xylan esterase in the solubilization of xylan and enzymatic hydrolysis of wheat straw and giant reed. Biotechnol Biofuels 4:60
Zheng F, Huang J, Yin Y (2013) A novel neutral xylanase with high SDS resistance from Volvariella volvacea: characterization and its synergistic hydrolysis of wheat bran with acetyl xylan esterase. J Ind Microbiol Biotechnol 40:1083–1093
Zhuo R, Yu H, Qin X et al (2018) Heterologous expression and characterization of a xylanase and xylosidase from white rot fungi and their application in synergistic hydrolysis of lignocellulose. Chemosphere 212:24–33
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no further conflicts of interest. The authors are responsible for the content and writing of this article and are grateful for financial support from the National Research Foundation (NRF) and Council for Scientific and Industrial Research (CSIR) in South Africa. Any opinion, findings and conclusions or recommendations expressed in this material are those of the author(s) and therefore the NRF does not accept any liability in regard thereto.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Malgas, S., Mafa, M.S., Mkabayi, L. et al. A mini review of xylanolytic enzymes with regards to their synergistic interactions during hetero-xylan degradation. World J Microbiol Biotechnol 35, 187 (2019). https://doi.org/10.1007/s11274-019-2765-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2765-z