
Abstract
Acetic acid is an inhibitor in industrial processes such as wine making and bioethanol production from cellulosic hydrolysate. It causes energy depletion, inhibition of metabolic enzyme activity, growth arrest and ethanol productivity losses in Saccharomyces cerevisiae. Therefore, understanding the mechanisms of the yeast responses to acetic acid stress is essential for improving acetic acid tolerance and ethanol production. Although 329 genes associated with acetic acid tolerance have been identified in the Saccharomyces genome and included in the database (http://www.yeastgenome.org/observable/resistance_to_acetic_acid/overview), the cellular mechanistic responses to acetic acid remain unclear in this organism. Post-genomic approaches such as transcriptomics, proteomics, metabolomics and chemogenomics are being applied to yeast and are providing insight into the mechanisms and interactions of genes, proteins and other components that together determine complex quantitative phenotypic traits such as acetic acid tolerance. This review focuses on these omics approaches in the response to acetic acid in S. cerevisiae. Additionally, several novel strains with improved acetic acid tolerance have been engineered by modifying key genes, and the application of these strains and recently acquired knowledge to industrial processes is also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic materials are rapidly becoming a major source of bioethanol because they are abundant, renewable, and uncompetitive with food resources. However, their pre-treatment usually generates many inhibitory compounds that hamper microorganism growth, such as formic acid, acetic acid and furfural (Fernandes et al. 2005; Luo et al. 2002). Since lignin and hemicellulose are highly acetylated, acetic acid is produced during the pre-treatment process, and it remains in the resultant hydrolysate (Chesson et al. 1983). Acetic acid also causes intracellular acidification, leading to energy depletion (Pampulha and Loureiro-Dias 2000) and inhibition of cell growth and metabolism during lignocellulosic fermentation. This weak acid is one of the main inhibitors in lignocellulosic hydrolysates.
Understanding the physiological mechanisms of Saccharomyces cerevisiae is a basic requirement for enhancing acetic acid tolerance during fermentation and improving product quality, such as balsamic vinegar and wine (Solieri and Giudici 2008; Vilela-Moura et al. 2011). Acetic acid tolerance is controlled by multiple genes, and even in a relatively simple model organism such as S. cerevisiae, precisely determining the genes involved and their interactions is highly challenging. Part of the reason is the difficulty in narrowing phenotypes to single genes and in detecting and quantifying epistasis, the contribution of variable quantitative traits and linked quantitative trait loci (Wilkening et al. 2014). Thus, the cellular stress responses to acetic acid remain unclear, which makes it difficult to improve this organism by genetic engineering.
After decades of post-genomic research, several approaches have been developed in S. cerevisiae at a genome-wide scale, including DNA microarray analysis, functional screening of non-essential gene deletion collections, whole genome sequence analysis, and inverse engineering. By using these technologies, we could dissect acetic acid tolerance and other complex traits in yeast. This review gives an overview of high-throughput technologies used to decipher acetic acid tolerance in S. cerevisiae. We discuss the advantages and challenges of these approaches, the novel genes identified, and novel strains constructed by modifying some key genes.
General mechanisms of acetic acid tolerance
In low pH conditions (<4.76), acetic acid is in an undissociated (protonated) state that can enter the cell through the Fps1p channel or by simple diffusion (Mollapour and Piper 2007). Once in the cytoplasm, acetic acid dissociates into acetate anions and protons, leading to cytoplasmic acidification and inhibition of some important metabolic processes (Arneborg et al. 2000). S. cerevisiae is sensitive to acetic acid in the presence of glucose, which restrains acetic acid depletion. Yeast can undergo programmed cell death triggered by internal and external stimuli including acetic acid (Giannattasio et al. 2013; Ludovico et al. 2001).
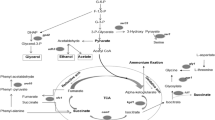
Nevertheless, yeast can survive in the presence of low concentrations of acetic acid. Acetate anions can be converted to acetyl-CoA by peroxisomal or cytosolic acetyl-CoA synthetases. Then acetyl-CoA enters the tricarboxylic cycle or glyoxylate cycle (Lee et al. 2011; Rolland et al. 2002; Vilela-Moura et al. 2008). Yeast adapted to acetic acid display many physiological alterations, including cell wall and membrane reorganization, pH recovery, efflux of anions, and detoxification (Mira et al. 2010c). Two important enzymes are required to maintain the pH in the cytoplasm: the plasma membrane proton-pumping ATPase (PM-H+-ATPase) and the vacuolar proton-pumping ATPase (V-H+-ATPase). PM-H+-ATPase is activated by weak acid, and pumps out protons in response (Carmelo et al. 1997). V-H+-ATPase also contributes to the recovery and maintenance of cytosolic pH, and it is crucial for the other physiological processes (Kane 2006; Martinez-Munoz and Kane 2008). Meanwhile, the ATP-binding cassette (ABC) transporter Pdr12 can export some organic anions out of the cell (Holyoak et al. 1999) (Fig. 1).

Mechanisms of acetic acid stress in S. cerevisiae. When yeast cells are in acetic acid environment, the cell wall structure will reconstruct and plasma membrane lipid composition will reconfigure. Yeast can utilize acetate anion through Jen1p or Ady2p when glucose doesn’t exist. At low pH, acetic acid enters cells in undissociated form by simple diffusion or through Fps1p. In the neutral cytosolic pH, acetic acid dissociated acetate anions and protons. Proton can be excluded out of the cytoplasm through Pma1p or V-ATPase to recover intracellular pH
Omics analysis of acetic acid tolerance
It is well known that most tolerance traits are controlled by multiple genetic loci, and omics analysis offers a promising approach for identifying the genetic basis of quantitative traits. Omics analysis can probe cells at a genome-wide scale to provide information on genome, transcriptome, proteome, and metabolome, due to the availability of complete genome sequences. Nowadays, more and more omics technologies are being used in the study of acetic acid tolerance in S. cerevisiae, and these are summarized below.
Functional genomics screening
With the development of high-throughput culturing technologies, many strains can be analysed to identify candidate genes involved in acetic acid tolerance. Gene deletion is the most common method deployed over the past 10 years. Kawahata et al. (2006) screened 4908 strains and identified genes involved in cell wall components that are important for responding to organic acids and low pH. Another group identified approximately 490 of the 650 determinants of tolerance to acetic acid by screening a ~5100 individuals from a haploid mutant collection for susceptibility phenotypes to acetic acid in non-essential deletion strains (Mira et al. 2010b). Candidate genes identified in these studies could be tested in genetic engineering experiments to obtain more robust strains.
Moreover, rapid molecular genetic manipulation technologies make it possible to screen strains overexpressing specific genes aimed at improving acetic acid tolerance. For example, Ma et al. (2015) screened strains transformed with an artificial zinc finger protein transcription factor (ZFP-TF) library and obtained strain ATCC4126 (Sc4126-MO1) that displayed improved acetic acid tolerance. They conducted further analysis and found three genes, YFL040W, QDR3, and IKS1, that are involved in the enhanced acetic acid tolerance, demonstrating the power of a synthetic ZFP-TF library for improving acetic acid tolerance. The employment of an artificial transcription factor can also facilitate the exploration of novel functional genes involved in stress tolerance. Functional genomes screening is performed by single gene deleted strains, but the essential genes are difficult to knock out. So, one drawback of this technique is the inconvenience of having to detect essential genes.
Genome analysis
In recent years, whole genome sequencing has substituted automated sanger sequencing for large-scale sequence data collection, mainly due to parallelization of the sequencing process. The available whole genome microarrays provide an enormous volume of inexpensive and accurate genome sequence data (Metzker 2010). By whole genome sequencing, four genes (ASG1, ADH3, SKS1 and GIS4) associated with acetic acid tolerance were identified (González-Ramos et al. 2016). Also, whole genome sequence analysis of pooled segregants can be widely used to identify genetic determinants throughout the genome (Ehrenreich et al. 2010), such as those involved in sporulation efficiency (Ben-Ari et al. 2006), ethanol tolerance (Swinnen et al. 2012a), maximal ethanol accumulation capacity (Pais et al. 2013), low glycerol production (Hubmann et al. 2013a, b), high thermotolerance (Yang et al. 2013), survival at low pH (Fletcher et al. 2015), and high acetic acid tolerance (Meijnen et al. 2016). In these studies, one or more segregants were sequenced at a whole genome level to dissect the relationships between phenotypes and genotypes. Additionally, this technology provides a new way of scoring large numbers of genetic markers distributed throughout the genome, such as single nucleotide polymorphisms (SNPs) (Otero et al. 2010) and simple sequence repeats (SSRs) (Geng et al. 2016). By combining bioinformatics with whole genome high-throughput sequencing, we have personally identified genes with specific SNPs that are associated with acetic acid tolerance (unpublished data).
Approaches for whole genome sequencing allow using relatively low numbers of selected segregants to identify major genetic loci. However, validation of this methodology through the identification of all causative genes within a genome remains a challenge. Furthermore, application to a large number of individual segregants remains time-consuming and costly (Swinnen et al. 2012b).
Transcriptome analysis
Genome-wide changes in transcription occur in yeast cells during the early stages of the response to acetic acid stress that are believed to be important as a first step in adapting to an acidic environment. Through genome-wide transcriptome analysis, a comprehensive view of acetic acid tolerance can be achieved. This technology could be further deployed to search for transcription regulon factors. Indeed, several studies have reported transcriptional changes in S. cerevisiae cells exposed to acetic acid. These include the following various types of such changes: (a) two short time (Li and Yuan 2010; Mira et al. 2010a); (b) two long time (Abbott et al. 2007; Bajwa et al. 2013); (c) one between short and long time (Lee et al. 2015); and (d) one both short and long time (Kawahata et al. 2006). By studying genomic expression using DNA microarray analysis, Haa1p was found to regulate, directly or indirectly, the transcription of approximately 80% of acetic acid-activated genes. This suggests that Haa1p is the main player controlling the yeast response to acetic acid (Mira et al. 2010a).
Transcriptome analysis can also identify genes that are up- or down-regulated in the presence of acetic acid. Ismail et al. (2014) compared the transcriptomes of cells supplemented with metal ion cultures and untreated controls, and found that up-regulation of FIT2, HXT1 and TKL1 could enhance xylose consumption in the presence of acetic acid. An et al. (2015) screened a library of mutants overexpressing alleles of the TATA-binding gene SPT15, and identified two S. cerevisiae strains with enhanced tolerance to acetic acid with 58 up-regulated genes and 106 down-regulated genes.
In recent years, RNA sequencing has emerged as a novel high-throughput approach for transcriptomic profiling based on deep-sequencing technologies (Braconi et al. 2016; Nagalakshmi et al. 2008). RNA sequencing approaches are particularly suitable for comparative analysis aimed at evaluating structural and gene expression variability among different strains and species (Sardu et al. 2014). By RNA sequencing, Chen et al. (2016a) identified 184 consensus genes between strain YC1 and S-C1 in response to the distinct inhibitor resistance and found key transcription factors that regulate these consensus genes. And as a generally applicable method, RNA interference (RNAi) can be used for genome-scale engineering. Si et al. (2015) showed that three rounds of iterative RNAi screening led to the identification of three gene knockdown targets that acted synergistically to confer an engineered yeast strain with substantially improved acetic acid tolerance. In the near future, RNA sequencing is likely to provide an invaluable contribution to environmental toxicological studies.
Proteomic analysis
Among the various methods, proteomic analysis provides a powerful way to analyse the cellular protein profile in the presence of acetic acid. Ghaemmaghami et al. (2003) created a fusion library of open reading frames tagged with a high-affinity epitope, making it possible to analyse the expression of misannotated genes at a global level. In acetic acid-induced programmed cell death progress, Longo et al. (2015) analysed proteomic data from wild type and Δyca1 cells and found that carbohydrate catabolism, lipid metabolism, proteolysis and stress responses were the main functional roles. Lv et al. (2014) analysed the effect of toxic compounds including acids, furans, and phenols on yeast. In proteomic analysis, 194 and 215 unique proteins were identified as differently expressed proteins at lag phase and exponential phase, respectively.
Metabolomic analysis
Metabolomics has emerged as an important tool in many disciplines (Cevallos-Cevallos et al. 2009). Generally, metabolite concentrations are the result of complicated cellular mechanisms, including both transcriptional and translational regulation. Therefore, metabolome data reflects the metabolic state of the cell better than transcriptomic or proteomic data. It is therefore superior for understanding what has happened and is happening inside a cell during a given biological process (Weckwerth 2003). Metabolomic analysis has been used to identify the key metabolites and metabolic reactions involved in stress and to improve stress tolerance (Ding et al. 2012; Hasunuma et al. 2011; Nugroho et al. 2015). An et al. (2015) used metabolome profile analysis to reveal that the intracellular concentrations of five metabolites were increased and 102 were decreased in S. cerevisiae MRRC3252. They also found that deletion of the urea degradation gene DUR1/2 and low levels of amino acids enhanced tolerance to acetic acid (An et al. 2015). Nugroho et al. (2015) conducted a metabolomics approach to investigate the effect of lactic acid-induced stress on metabolite pools in S. cerevisiae. And their results suggested that the addition of proline improves the specific growth rate and protects cells from acid stress by combating acid-induced oxidative stress.
Genetic manipulation for improving acetic acid tolerance
Omics approaches could provide the theoretical basis and definite genes for genetic modification in yeast. By functional genomics screening and genome wide analysis, many genes associated with acetic acid tolerance are involved in carbohydrate metabolism, protein folding, lipid metabolism, cell wall function and transport (Mira et al. 2010b). There are several examples of genetic manipulation to alter the cell structure and thereby improve acetic acid tolerance.
The plasma membrane is an important barrier in acetic acid tolerance. When acetic acid is present, the expression of YRO2 is induced in the plasma membrane, and the Yro2 protein is believed to alleviate acetic acid-induced damage (Takabatake et al. 2015). Acetic acid can also enter the cell through the Fps1p channel. While deletion of fps1 can improve ethanol production and decrease acetic acid yield. Part of the reason was that the fps1∆ mutant might solve the occurring redox balance problems by reducing acetic acid (Zhang et al. 2007).
In the cytoplasm, acetic acid dissociates into acetate anions and protons, and the consumption of acetate and/or protection of intracellular proteins can improve acetic acid tolerance. Through genomic library screening, Chen et al. (2016b) found that the endogenous expression of Whi2 could be activated by acetic acid. So, overexpression of WHI2, encoding a 55 kDa cytoplasmic globular scaffold protein, can improve acetic acid tolerance and provide a protective effect in S. cerevisiae. And overexpression of ACS2, an acetyl-coenzyme A synthetase, also improves resistance to acetic acid during fermentation (Ding et al. 2015b). Wu et al. (2016) found that higher long chain fatty acid concentrations, intracellular trehalose levels, and CAT activity are important factors contributing to the improved acetic acid tolerance of jjj1∆ mutants.
Mitochondria are the epicentre of energy production in the cell, and an important organelle for tolerance to acetic acid. Kumar et al. (2015) overexpressed COX20, a mitochondrial cytochrome c oxidase chaperone, which improved tolerance to acetic acid during fermentation.
The biogenesis and fragmentation of the vacuole is dependent on vesicle–vesicle and vesicle–vacuole fusion and fission events. By screening overexpression library, overexpression of PEP3, VAM6, or VPS3 increases the number of fragmented vacuoles, and overexpression of PEP3 in particular shortens the lag phase associated with stress tolerance without altering the growth rate (Ding et al. 2015a).
The nucleus also plays a role in acetic acid tolerance. Chromatin dynamics controlled by various histone modification enzymes is important for stress-responsive genes, and histone H3/H4 acetylation is an important histone modification. By screening 345 single point mutations in H3 and H4 histone libraries, Liu et al. (2014) identified two mutants, H3K37A and H4K16Q, that showed improved ethanol fermentation ability under acetic acid stress conditions. And they further analysed acetic acid tolerance and ethanol fermentation capacity of the mutants by genome-wide transcriptional profiling. As we know, RTT109 is a histone acetyltransferase for the acetylation of histone H3. Cheng et al. (2016) fought that the absence of RTT109 not only activates transcription of stress responsive genes, but also improves resistance to oxidative stress, which ultimately contributes to improved acetic acid tolerance in S. cerevisiae.
To date, many studies have focused on laboratory strains, but few have investigated industrial strains or naturally occurring organisms isolated from the environment. This may be because industrial strains have a more complex genome, since they are often diploid, polyploid, or aneuploid. In general, a true and stable haploid strain is difficult to obtain. Furthermore, phenotypic screening can be laborious and time-consuming, hence it is difficult to directly identify some industrially relevant traits using bulk segregant approaches (Swinnen et al. 2012b). However, several successful strategies using high-throughput technologies can now be applied to industrial strains. In one study, highly ethanol tolerant industrial strains were investigated and three causative genes (MKT1, SKS2 and APJ1) identified by pooled-segregant whole genome sequence analysis (Swinnen et al. 2012a). Another study crossed haploid segregants of a strain with an unusually high acetic acid tolerance and a reference industrial strain, resulting in the identification of three new causative alleles (GLO1, DOT5, CUP2) that determine high acetic acid tolerance (Meijnen et al. 2016). Such methods are therefore capable of dissecting specific phenotypes at a whole genome scale, and the knowledge obtained can be used to improve acetic acid tolerance in industrial strains (Table 1).
Perspectives
Several molecular factors have been identified that are related to acetic acid and modified to engineer improved strains. These include strains overexpressing HAA1 and GRX5 (Fang et al. 2015; Sakihama et al. 2015; Swinnen et al. 2017), strains in which FPS1 and PHO13 are deleted (Sakihama et al. 2015), and 52 individual deletion strains identified by screening a deletion mutant library. However, because of the complex mechanisms involved in the response to acetic acid in yeast, a global molecular analysis of strains by whole genome sequencing and/or omics technologies is required to assess weak acid tolerance. Fortunately, recent advances in high-throughput techniques, including genomics, transcriptomics, proteomics, and metabolomics, are providing us with a vast amount of biological data to help us unravel these biological processes. Furthermore, these results can be used to engineer more robust strains with improved stress resistance.
Whole genome sequencing can certainly initiate a better understanding of stress tolerance, but it cannot distinguish between trait-relevant and trait-irrelevant molecular differences. Bansal (2005) summarized the limitations in genomics and proteomics approaches as follows: a lack of available gene-functionality information in wet-lab data, a lack of computer algorithms to explore the vast amount of data with unknown functionality, a limited knowledge of protein–protein and protein–DNA interactions, and a lack of knowledge concerning the temporal and transient behaviour of genes and pathways. Interestingly, genome-wide profiles are rarely in agreement, apparently due to different experimental conditions, including the strains used, the pH of the medium, and the acetic acid concentration. However, all data are valuable for both reinforcing previous findings and understanding regulatory networks (Mira et al. 2010c). Further elucidation of the molecular mechanisms of acetic acid tolerance will likely require a combination of high-throughput approaches and other methods, such as gene manipulation techniques including CRISPER/Cas9, systems metabolic analysis and evolution engineering.
References
Abbott DA, Knijnenburg TA, de Poorter LMI, Reinders MJT, Pronk JT, van Maris AJA (2007) Generic and specific transcriptional responses to different weak organic acids in anaerobic chemostat cultures of Saccharomyces cerevisiae. FEMS Yeast Res 7:819–833. doi:10.1111/j.1567-1364.2007.00242.x
An J, Kwon H, Kim E, Lee YM, Ko HJ, Park H, Choi IG, Kim S, Kim KH, Kim W, Choi W (2015) Tolerance to acetic acid is improved by mutations of the TATA-binding protein gene. Environ Microbiol 17:656–669. doi:10.1111/1462-2920.12489
Arneborg N, Jespersen L, Jakobsen M (2000) Individual cells of Saccharomyces cerevisiae and Zygosaccharomyces bailii exhibit different short-term intracellular pH responses to acetic acid. Arch Microbiol 174:125–128. doi:10.1007/s002030000185
Bajwa PK, Ho CY, Chan CK, Martin VJJ, Trevors JT, Lee H (2013) Transcriptional profiling of Saccharomyces cerevisiae T2 cells upon exposure to hardwood spent sulphite liquor: comparison to acetic acid, furfural and hydroxymethylfurfural. Antonie Van Leeuwenhoek 103:1281–1295. doi:10.1007/s10482-013-9909-1
Bansal AK (2005) Bioinformatics in microbial biotechnology—a mini review. Microb Cell Fact 4:11. doi:10.1186/1475-2859-4-19
Ben-Ari G, Zenvirth D, Sherman A, David L, Klutstein M, Lavi U, Hillel J, Simchen G (2006) Four linked genes participate in controlling sporulation efficiency in budding yeast. PLoS Genet 2:1815–1823. doi:10.1371/journal.pgen.0020195
Braconi D, Bernardini G, Santucci A (2016) Saccharomyces cerevisiae as a model in ecotoxicological studies: a post-genomics perspective. J Proteomics 137:19–34. doi:10.1016/j.jprot.2015.09.001
Carmelo V, Santos H, Sá-Correia I (1997) Effect of extracellular acidification on the activity of plasma membrane ATPase and on the cytosolic and vacuolar pH of Saccharomyces cerevisiae. Biochim Biophys Acta 1325(1):63–70. doi:10.1016/s0005-2736(96)00245-3
Cevallos-Cevallos JM, Reyes-De-Corcuera JI, Etxeberria E, Danyluk MD, Rodrick GE (2009) Metabolomic analysis in food science: a review. Trends Food Sci Technol 20:557–566 doi:10.1016/j.tifs.2009.07.002
Chen YY, Sheng JY, Jiang T, Stevens J, Feng XY, Wei N (2016a) Transcriptional profiling reveals molecular basis and novel genetic targets for improved resistance to multiple fermentation inhibitors in Saccharomyces cerevisiae. Biotechnol Biofuels 9:18. doi:10.1186/s13068-015-0418-5
Chen YY, Stabryla L, Wei N (2016b) Improved acetic acid resistance in Saccharomyces cerevisiae by overexpression of the WHI2 gene identified through inverse metabolic engineering. Appl Environ Microbiol 82:2156–2166. doi:10.1128/aem.03718-15
Cheng C, Zhao XQ, Zhang MM, Bai FW (2016) Absence of Rtt109p, a fungal-specific histone acetyltransferase, results in improved acetic acid tolerance of Saccharomyces cerevisiae. FEMS Yeast Res 16:9. doi:10.1093/femsyr/fow010
Chesson A, Gordon AH, Lomax JA (1983) Substituent groups linked by alkali-labile bonds to arabinose and xylose residues of legume, grass and cereal straw cell walls and their fate during digestion by rumen microorganisms. J Sci Food Agric 34:1330–1340. doi:10.1002/jsfa.2740341204
Ding MZ, Wang X, Yang Y, Yuan YJ (2012) Comparative metabolic profiling of parental and inhibitors-tolerant yeasts during lignocellulosic ethanol fermentation. Metabolomics 8:232–243. doi:10.1007/s11306-011-0303-6
Ding J, Holzwarth G, Bradford CS, Cooley B, Yoshinaga AS, Patton-Vogt J, Abeliovich H, Penner MH, Bakalinsky AT (2015a) PEP3 overexpression shortens lag phase but does not alter growth rate in Saccharomyces cerevisiae exposed to acetic acid stress. Appl Microbiol Biotechnol 99:8667–8680. doi:10.1007/s00253-015-6708-9
Ding J, Holzwarth G, Penner MH, Patton-Vogt J, Bakalinsky AT (2015b) Overexpression of acetyl-CoA synthetase in Saccharomyces cerevisiae increases acetic acid tolerance. FEMS Microbiol Lett 362:7. doi:10.1093/femsle/fnu042
Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L (2010) Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature 464:1039–1101. doi:10.1038/nature08923
Fang Q, Zhang M, Chen H, Xiong L, Zhao X, Bai F (2015) Improvement of acetic acid tolerance of Saccharomyces cerevisiae by overexpressing glutaredoxin encoding gene GRX5. CIESC J 66:1434–1439
Fernandes AR, Mira NP, Vargas RC, Canelhas I, Sa-Correia I (2005) Saccharomyces cerevisiae adaptation to weak acids involves the transcription factor Haa1p and Haa1p-regulated genes. Biochem Biophys Res Commun 337:95–103. doi:10.1016/j.bbrc.2005.09.010
Fletcher E, Feizi A, Kim S, Siewers V, Nielsen J (2015) RNA-seq analysis of Pichia anomala reveals important mechanisms required for survival at low pH. Microb Cell Fact 14:11. doi:10.1186/s12934-015-0331-4
Geng P, Xiao Y, Hu Y, Sun HY, Xue W, Zhang L, Shi GY (2016) Genetic dissection of acetic acid tolerance in Saccharomyces cerevisiae. World J Microbiol Biotechnol 32:8. doi:10.1007/s11274-016-2101-9
Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425:737–741. doi:10.1038/nature02046
Giannattasio S, Guaragnella N, Zdralevic M, Marra E (2013) Molecular mechanisms of Saccharomyces cerevisiae stress adaptation and programmed cell death in response to acetic acid Frontiers. Microbiology 4:7. doi:10.3389/fmicb.2013.00033
González-Ramos D, Gorter de Vries AR, Grijseels SS, van Berkum MC, Swinnen S, van den Broek M, Nevoigt E, Daran JM, Pronk JT, van Maris AJ (2016) A new laboratory evolution approach to select for constitutive acetic acid tolerance in Saccharomyces cerevisiae and identification of causal mutations. Biotechnol Biofuels 9:18 doi:10.1186/s13068-016-0583-1
Hasunuma T, Sanda T, Yamada R, Yoshimura K, Ishii J, Kondo A (2011) Metabolic pathway engineering based on metabolomics confers acetic and formic acid tolerance to a recombinant xylose-fermenting strain of Saccharomyces cerevisiae. Microb Cell Fact 10:13. doi:10.1186/1475-2859-10-2
Holyoak CD, Bracey D, Piper PW, Kuchler K, Coote PJ (1999) The Saccharomyces cerevisiae weak-acid-inducible ABC transporter pdr12 transports fluorescein and preservative anions from the cytosol by an energy-dependent mechanism. J Bacteriol 181:4644–4652
Hubmann G, Foulquié-Moreno MR, Nevoigt E, Duitama J, Meurens N, Pais TM, Mathé L, Saerens S, Nguyen HT, Swinnen S, Verstrepen KJ, Concilio L, de Troostembergh JC, Thevelein JM (2013a) Quantitative trait analysis of yeast biodiversity yields novel gene tools for metabolic engineering. Metab Eng 17:68–81. doi:10.1016/j.ymben.2013.02.006
Hubmann G, Mathé L, Foulquié-Moreno MR, Duitama J, Nevoigt E, Thevelein JM (2013b) Identification of multiple interacting alleles conferring low glycerol and high ethanol yield in Saccharomyces cerevisiae ethanolic fermentation. Biotechnol Biofuels 6(1):87 doi:10.1186/1754-6834-6-87
Ismail KSK, Sakamoto T, Hasunuma T, Zhao XQ, Kondo A (2014) Zinc, magnesium, and calcium ion supplementation confers tolerance to acetic acid stress in industrial Saccharomyces cerevisiae utilizing xylose. Biotechnol J 9:1519–1525. doi:10.1002/biot.201300553
Kane PA (2006) The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol Mol Biol Rev 70:177–191. doi:10.1128/mmbr.70.1.177-191.2006
Kawahata M, Masaki K, Fujii T, Iefuji H (2006) Yeast genes involved in response to lactic acid and acetic acid: acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res 6:924–936. doi:10.1111/j.1567-1364.2006.00089.x
Kim SK, Jin YS, Choi IG, Park YC, Seo JH (2015) Enhanced tolerance of Saccharomyces cerevisiae to multiple lignocellulose-derived inhibitors through modulation of spermidine contents. Metab Eng 29:46–55. doi:10.1016/j.ymben.2015.02.004
Kumar V, Hart AJ, Keerthiraju ER, Waldron PR, Tucker GA, Greetham D (2015) Expression of mitochondrial cytochrome c oxidase chaperone gene (COX20) improves tolerance to weak acid and oxidative stress during yeast fermentation. PLoS ONE 10:16. doi:10.1371/journal.pone.0139129
Lee YJ, Jang JW, Kim KJ, Maeng PJ (2011) TCA cycle-independent acetate metabolism via the glyoxylate cycle in Saccharomyces cerevisiae. Yeast 28:153–166. doi:10.1002/yea.1828
Lee Y, Nasution O, Choi E, Choi IG, Kim W, Choi W (2015) Transcriptome analysis of acetic-acid-treated yeast cells identifies a large set of genes whose overexpression or deletion enhances acetic acid tolerance. Appl Microbiol Biotechnol 99:6391–6403. doi:10.1007/s00253-015-6706-y
Li B-Z, Yuan Y-J (2010) Transcriptome shifts in response to furfural and acetic acid in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 86:1915–1924. doi:10.1007/s00253-010-2518-2
Liu XY, Zhang XH, Zhang ZJ (2014) Point mutation of H3/H4 histones affects acetic acid tolerance in Saccharomyces cerevisiae. J Biotechnol 187:116–123. doi:10.1016/j.jbiotec.2014.07.445
Longo V, Ždralević M, Guaragnella N, Giannattasio S, Zolla L, Timperio AM (2015) Proteome and metabolome profiling of wild-type and YCA1-knock-out yeast cells during acetic acid-induced programmed cell death. J Proteomics 128:173–188. doi:10.1016/j.jprot.2015.08.003
Ludovico P, Sousa MJ, Silva MT, Leão C, Côrte-Real M (2001) Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 147:2409–2415. doi:10.1099/00221287-147-9-2409
Luo CD, Brink DL, Blanch HW (2002) Identification of potential fermentation inhibitors in conversion of hybrid poplar hydrolyzate to ethanol. Biomass Bioenergy 22:125–138 doi:10.1016/s0961-9534(01)00061-7
Lv YJ, Wang X, Ma Q, Bai X, Li BZ, Zhang WW, Yuan YJ (2014) Proteomic analysis reveals complex metabolic regulation in Saccharomyces cerevisiae cells against multiple inhibitors stress. Appl Microbiol Biotechnol 98:2207–2221. doi:10.1007/s00253-014-5519-8
Ma C, Wei XW, Sun CH, Zhang F, Xu JR, Zhao XQ, Bai FW (2015) Improvement of acetic acid tolerance of Saccharomyces cerevisiae using a zinc-finger-based artificial transcription factor and identification of novel genes involved in acetic acid tolerance. Appl Microbiol Biotechnol 99:2441–2449. doi:10.1007/s00253-014-6343-x
Martinez-Munoz GA, Kane P (2008) Vacuolar and plasma membrane proton pumps collaborate to achieve cytosolic pH homeostasis in yeast. J Biol Chem 283:20309–20319. doi:10.1074/jbc.M710470200
Meijnen JP, Randazzo P, Foulquié-Moreno MR, van den Brink J, Vandecruys P, Stojiljkovic M, Dumortier F, Zalar P, Boekhout T, Gunde-Cimerman N, Kokošar J, Štajdohar M, Curk T, Petrovič U, Thevelein JM (2016) Polygenic analysis and targeted improvement of the complex trait of high acetic acid tolerance in the yeast Saccharomyces cerevisiae. Biotechnol Biofuels. doi:10.1186/s13068-015-0421-x
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11:31–46. doi:10.1038/nrg2626
Mira NP, Becker JD, Sá-Correia I (2010a) Genomic expression program involving the Haa1p-regulon in Saccharomyces cerevisiae response to acetic acid Omics-a. J Integr Biol 14:587–601. doi:10.1089/omi.2010.0048
Mira NP, Palma M, Guerreiro JF, Sá-Correia I (2010b) Genome-wide identification of Saccharomyces cerevisiae genes required for tolerance to acetic acid. Microb Cell Fact 9:13. doi:10.1186/1475-2859-9-79
Mira NP, Teixeira MC, Sá-Correia I (2010c) Adaptive response and tolerance to weak acids in Saccharomyces cerevisiae: a genome-wide view Omics-a. J Integr Biol 14:525–540. doi:10.1089/omi.2010.0072
Mollapour M, Piper PW (2007) Hog1 mitogen-activated protein kinase phosphorylation targets the yeast Fps1 aquaglyceroporin for endocytosis, thereby rendering cells resistant to acetic acid. Mol Cell Biol 27:6446–6456. doi:10.1128/mcb.02205-06
Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320:1344–1349. doi:10.1126/science.1158441
Nugroho RH, Yoshikawa K, Shimizu H (2015) Metabolomic analysis of acid stress response in Saccharomyces cerevisiae. J Biosci Bioeng 120:396–404. doi:10.1016/j.jbiosc.2015.02.011
Otero JM, Vongsangnak W, Asadollahi MA, Olivares-Hernandes R, Maury J, Farinelli L, Barlocher L, Osterås M, Schalk M, Clark A, Nielsen J (2010) Whole genome sequencing of Saccharomyces cerevisiae: from genotype to phenotype for improved metabolic engineering applications. BMC Genomics 11:723. doi:10.1186/1471-2164-11-723
Pais TM, Foulquié-Moreno MR, Hubmann G, Duitama J, Swinnen S, Goovaerts A, Yang Y, Dumortier F, Thevelein JM (2013) Comparative polygenic analysis of maximal ethanol accumulation capacity and tolerance to high ethanol levels of cell proliferation in yeast. PLoS Genet 9:18. doi:10.1371/journal.pgen.1003548
Pampulha ME, Loureiro-Dias MC (2000) Energetics of the effect of acetic acid on growth of Saccharomyces cerevisiae. FEMS Microbiol Lett 184:69–72. doi:10.1016/s0378-1097(00)00022-7
Rolland F, Winderickx J, Thevelein JM (2002) Glucose-sensing and -signalling mechanisms in yeast. FEMS Yeast Res 2:183–201. doi:10.1111/j.1567-1364.2002.tb00084.x
Sakihama Y, Hasunuma T, Kondo A (2015) Improved ethanol production from xylose in the presence of acetic acid by the overexpression of the HAA1 gene in Saccharomyces cerevisiae. J Biosci Bioeng 119:297–302. doi:10.1016/j.jbiosc.2014.09.004
Sardu A, Treu L, Campanaro S (2014) Transcriptome structure variability in Saccharomyces cerevisiae strains determined with a newly developed assembly software. BMC Genomics. doi:10.1186/1471-2164-15-1045
Si T, Luo YZ, Bao ZH, Zhao HM (2015) RNAi-assisted genome evolution in Saccharomyces cerevisiae for complex phenotype engineering. ACS Synth Biol 4:283–291. doi:10.1021/sb500074a
Solieri L, Giudici P (2008) Yeasts associated to traditional balsamic vinegar: ecological and technological features. Int J Food Microbiol 125:36–45. doi:10.1016/j.ijfoodmicro.2007.06.022
Swinnen S, Schaerlaekens K, Pais T, Claesen J, Hubmann G, Yang Y, Demeke M, Foulquié-Moreno MR, Goovaerts A, Souvereyns K, Clement L, Dumortier F, Thevelein JM (2012a) Identification of novel causative genes determining the complex trait of high ethanol tolerance in yeast using pooled-segregant whole-genome sequence analysis. Genome Res 22:975–984. doi:10.1101/gr.131698.111
Swinnen S, Thevelein JM, Nevoigt E (2012b) Genetic mapping of quantitative phenotypic traits in Saccharomyces cerevisiae. FEMS Yeast Res 12:215–227. doi:10.1111/j.1567-1364.2011.00777.x
Swinnen S, Henriques SF, Shrestha R, Ho PW, Sá-Correia I, Nevoigt E (2017) Improvement of yeast tolerance to acetic acid through Haa1 transcription factor engineering: towards the underlying mechanisms. Microb Cell Fact 16:15. doi:10.1186/s12934-016-0621-5
Takabatake A, Kawazoe N, Izawa S (2015) Plasma membrane proteins Yro2 and Mrh1 are required for acetic acid tolerance in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 99:2805–2814. doi:10.1007/s00253-014-6278-2
Vilela-Moura A, Schuller D, Mendes-Faia A, Corte-Real M (2008) Reduction of volatile acidity of wines by selected yeast strains. Appl Microbiol Biotechnol 80:881–890. doi:10.1007/s00253-008-1616-x
Vilela-Moura A, Schuller D, Mendes-Faia A, Silva RD, Chaves SR, Sousa MJ, Côrte-Real M (2011) The impact of acetate metabolism on yeast fermentative performance and wine quality: reduction of volatile acidity of grape musts and wines. Appl Microbiol Biotechnol 89:271–280. doi:10.1007/s00253-010-2898-3
Weckwerth W (2003) Metabolomics in systems biology. Annu Rev Plant Biol 54:669–689. doi:10.1146/annurev.arplant.54.031902.135014
Wilkening S, Lin G, Fritsch ES, Tekkedil MM, Anders S, Kuehn R, Nguyen M, Aiyar RS, Proctor M, Sakhanenko NA, Galas DJ, Gagneur J, Deutschbauer A, Steinmetz LM (2014) An evaluation of high-throughput approaches to QTL mapping in Saccharomyces cerevisiae. Genetics 196:853–865. doi:10.1534/genetics.113.160291
Wu XC, Zhang LJ, Jin XN, Fang YH, Zhang K, Qi L, Zheng DQ (2016) Deletion of JJJ1 improves acetic acid tolerance and bioethanol fermentation performance of Saccharomyces cerevisiae strains. Biotechnol Lett 38:1097–1106. doi:10.1007/s10529-016-2085-4
Yang Y, Foulquié-Moreno MR, Clement L, Erdei E, Tanghe A, Schaerlaekens K, Dumortier F, Thevelein JM (2013) QTL analysis of high thermotolerance with superior and downgraded parental yeast strains reveals new minor QTLs and converges on novel causative alleles involved in RNA processing. PLoS Genet 9:15. doi:10.1371/journal.pgen.1003693
Zhang A, Kong Q, Cao L, Chen X (2007) Effect of FPS1 deletion on the fermentation properties of Saccharomyces cerevisiae. Lett Appl Microbiol 44:212–217. doi:10.1111/j.1472-765X.2006.02041.x
Acknowledgements
This work was funded by China Spark Program (Grant No. 2015GA690004) and the Outstanding Youth Foundation of Jiangsu Province (Grant No. BK20140002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Geng, P., Zhang, L. & Shi, G.Y. Omics analysis of acetic acid tolerance in Saccharomyces cerevisiae . World J Microbiol Biotechnol 33, 94 (2017). https://doi.org/10.1007/s11274-017-2259-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-017-2259-9