
Abstract
In this study, we developed a novel technique for preparing polyvinyl alcohol (PVA) hydrogel as an immobilizing matrix by the addition of sodium bicarbonate. This resulted in an increase in the specific surface area of PVA_sodium bicarbonate (PVA_SB) hydrogel beads to 65.23 m2 g−1 hydrogel beads, which was approximately 85 and 14 % higher than those of normal PVA and PVA_sodium alginate (PVA_SA) hydrogel beads, respectively. The D e value of PVA_SB hydrogel beads was calculated as 7.49 × 10−4 cm2 s−1, which was similar to the D e of PVA_SA hydrogel beads but nearly 38 % higher than that of the normal PVA hydrogel beads. After immobilization with nitrifying biomass, the oxygen uptake rate and the ammonium oxidation rate of nitrifying biomass entrapped in PVA_SB hydrogel beads were determined to be 19.53 mg O2 g MLVSS−1 h−1 and 10.59 mg N g MLVSS−1 h−1, which were 49 and 43 % higher than those of normal PVA hydrogel beads, respectively. Scanning electron microscopy observation of the PVA_SB hydrogel beads demonstrated relatively higher specific surface area and revealed loose microstructure that was considered to provide large spaces for microbial growth. This kind of structure was also considered beneficial for reducing mass transfer resistance and increasing pollutant uptake.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyvinyl alcohol (PVA) possesses useful chemical characteristics such as hydrophilicity, relatively large surface area and resistance to oxidation. In particular, PVA hydrogel beads are harmless and nontoxic to biomass, so PVA is a suitable carrier material to immobilize biomass for different application. Due to these properties mentioned above, PVA has been applied in many aspects, such as the fermentation process (Bezbradica et al. 2007), food industry (Lei et al. 2011; Tripathi et al. 2009), medicine industry (Puppi et al. 2011), chemical engineering industry (Chirizzi et al. 2011), and environmental engineering processes (Quan et al. 2011; Sandeman et al. 2011). Successful application of immobilized microorganisms has also been documented for the removal of contaminants such as organic carbon, nitrogen, dye, and phenol (Chou et al. 2010; El-Naas et al. 2009; Khan and Banerjee 2010).
Although entrapment of microorganisms in PVA hydrogel beads have many advantages compared to other immobilizing techniques, the entrapped microorganisms in the carriers by PVA-boric acid method (Hashimoto and Furukawa 1987) and freeze-thawing method (Ariga et al. 1987; Ricciardi et al. 2004) might suffer severe mass transfer resistance, for example hindrance of substrate diffusion to the immobilized microorganisms and metabolites from the carrier. Khoo and Ting (2001) prepared PVA calcium alginate beads to entrap fungal biomass for heavy metal uptake. They found that addition of alginate resulted in larger bore diameter compared to those without alginate addition, which further provided a relatively lower mass transfer resistance. Wu and Wisecarver (1992) suggested that addition of sodium alginate (SA) with concentration below 0.4 % (w/v) could prevent the agglomeration of PVA hydrogel beads, but there was no effect on the improvement of PVA network structure formation. Wang et al. (2006) reported that the addition of SA with concentration about 1.0 % (w/v) could not only avoid the agglomeration of PVA hydrogel beads but also efficiently improve and control PVA network structure formation. Wang et al. (2007) utilized the complex of PVA and SA crosslinked with Ca(NO3)2, which was proved to be more suitable for entrapping Acidithiobacillus ferrooxidans. This entrapment technique achieved maximum iron oxidation rate of 4.6 g l−1 h−1 in batch culture; while 96 % of the initial ferrous iron was oxidized within 2.5 h in a packed-bed reactor for long term experiments.
In this study, we developed a novel technique for the preparation of PVA biomass carrier by adding sodium bicarbonate (SB) to PVA solution to form PVA_sodium bicarbonate (PVA_SB) hydrogel beads. The main aims of this manuscript are as follows: (1) to compare physical properties of PVA_SB hydrogel beads to those of normal PVA and modified PVA_sodium alginate (PVA_SA) hydrogel beads, including specific surface area, mass transfer rate, and oxygen uptake rate (OUR); (2) to explore the difference of structure of the three kinds of PVA hydrogel beads by scanning electron microscopy (SEM). Nitrifying biomass was used as test microorganisms in order to investigate the suitability of the carrier for microbial immobilization.
Materials and methods
Raw materials and cultivation of microorganisms
Polyvinyl alcohol, with polymerization degree of 1750 and alcoholysis degree higher than 99 %, was procured from Sinopharm Chemical Group Co., Ltd. (China). Sodium bicarbonate, sodium alginate, and other chemicals were all analytical reagent grade obtained from Beijing Chemical Reagent Factory (China).
Nitrifying biomass was taken from one nitrification tank in the Dalian Lingshui Municipal Wastewater Treatment Plant (Dalian, China). Before the immobilization experiments, the nitrifying biomass was cultivated for about 2 months in a laboratory-scale biofilm continuous reactor, and the hydraulic retention time was 8.5 h. The nitrifying biomass had a total nitrogen (TN) conversion rate of 1,400 g N m−3 day−1 with effluent nitrite over 90 % and nitrate content below 10 % as determined during 60 days of operation prior to immobilization. The composition of the mineral medium and the pH of the synthetic wastewater were as specified by Qiao et al. (2010), which mainly utilized NH4HCO3 as N source (100–500 mg/l as N). Other compounds were also included as following (g l−1): NaCl 1.0, CaCl2·2H2O 0.07, FeSO4·7H2O 0.009, KH2PO4·0.07, MgSO4·7H2O 0.05 and 1.5 ml l−1 of a trace element solution. The detailed components and concentrations of trace element solution were shown in Table 1. Samples (1 ml) were withdrawn every 3 days for the determination of the ammonium oxidation activity.
Preparation of PVA hydrogel carriers
Three types of PVA hydrogel beads were prepared using conventional PVA, PVA_SA and our newly developed PVA_SB technique. The preparation process for conventional PVA hydrogel beads was similar to that described by Quan et al. (2011) except that the solidifying solution was changed to 3 % boric acid and 2 % CaCl2, and the pH adjusted to 7.0 by 2 M NaOH. 7.5 % PVA (w/v) was firstly heated at 120° for 20 min by autoclave and then mixed uniformly. The mixture was cooled to about 35 °C and dropped slowly into the solidifying solution, which was continuously mixed on a magnetic stirrer to form spherical hydrogel beads, by a peristaltic pump (BT100-2J, Lange, Baoding, China). The PVA hydrogel beads were kept immersed in the solidifying solution for 12 h at room temperature to enhance their mechanical strength. The PVA hydrogel beads were finally washed with distilled water several times and used for the following experiments. The PVA_SA hydrogel beads were prepared in the same flow with the exception that 1.0 % (w/v) SA was added to 7.5 % (w/v) PVA concentration.
For the preparation of PVA_SB hydrogel beads, 0.25 % SB (w/v) was added to the dissolved PVA mixture as it cooled down to about 35 °C after heating at 120 °C. 6 M hydrochloric acid was added to the solution and the mixture was stirred vigorously to produce CO2 gas bubbles. The mixture was poured into the solidifying solution followed by the completion of the remaining steps as described above.
For microorganism entrapment, 5.0 g nitrifying biomass [wet weight, volatile suspended solids (VSS) 0.221 g] was added into each of the PVA solution, PVA_SA solution or PVA_SB solution when the solution temperature decreased to about 35 °C. The final percentages of PVA and nitrifying biomass were 7.5 and 0.11 % (w/v), respectively in each of the three kinds of hydrogel beads; while the final percentages of SA and SB were 2.0 and 0.25 % (w/v) in PVA_SA and PVA_SB hydrogel beads, respectively.
Oxygen uptake rate (OUR) of entrapped nitrifying biomass
Oxygen uptake rate was determined according to method described by Surmacz-Gorska et al. (1996). Around 5.0 g (wet weight) of entrapped PVA hydrogel carriers with nitrifying biomass (VSS 0.221 g) were added to a flask containing 200 ml synthetic wastewater, which was aerated for about 5 min to saturate the experiment solution with dissolved oxygen (DO) at room temperature about 28–30 °C. Aeration was then stopped and the DO values were immediately monitored every minute using a DO meter (YSI, Model 55, USA). OUR [mgO2 g mixed liquor volatile suspended solids (MLVSS)−1 h−1] was calculated from the slope of DO concentration. The OUR experiments with each kind of PVA hydrogel bead were done in triplicate.
Ammonium oxidation activity of entrapped nitrifying biomass
Approximately 0.221 g (dry weight) nitrifying biomass was entrapped into PVA beads and immersed in 400 ml synthetic wastewater (the same composition as that of the continuous reactor except the ammonium concentration), which was constantly mixed at 500 rev min−1 on a magnetic stirrer at room temperature (about 28–30 °C). The initial ammonium concentration in the media was set at 100 mg l−1 and the initial pH was adjusted to 7.8 and was not controlled during the ensuing experiment. Samples (1 ml) were withdrawn at regular intervals for the determination of the ammonium oxidation activity. The activity was expressed as specific ammonium oxidizing rate expressed as mg N g−1 MLVSS h−1. The ammonium oxidation activity experiments with each kind of PVA hydrogel bead were also done in triplicate.
Characterization of PVA hydrogel beads
The specific surface areas of the three types of hydrogel beads were determined using the adsorption procedure described by Yu et al. (2001), Wang et al. (2005) and Bai et al. (2010). The methylene blue (MB) stock solution used in this characterization had a concentration of 100 mg l−1 and this solution was used to prepare dilutions. Adsorption experiments were performed in 200 ml containers with 100 ml of the MB solution of different concentrations. 1.0 g hydrogel carrier beads were added into these containers with 100 ml MB solutions of various concentrations (10–90 mg l−1). The MB concentration in the solution was determined by a spectrophotometer (V-560, Jasco, Japan) at the wavelength of 665 nm. The samples were placed on a magnetic heating mixer (JB-5, Jintan Instruments Co., Ltd., Shenglan, China) maintained at a constant temperature of 30 °C during the whole experiments. A modified method of measuring specific surface area was used based on the theory of single layer molecular absorption of MB on solid surfaces (Bai et al. 2010; Hang and Brindley 1970).
The mass of adsorbed MB in the monolayer can be represented by Langmuir equation:
where Ce is the concentration of MB solution at adsorption equilibrium (g MB l−1); b is the adsorption constant (l g−1); Q is the adsorbed MB at adsorption equilibrium (g MB g−1 carrier); and Q max is the maximum mass of MB adsorbed on the monolayer (g MB g−1 carrier). Q max can be obtained from the reciprocal of the linear regression, and the diffusivity coefficient can be calculated from Q max (mg MB g−1 carrier) multiplied by 2.45.
The adsorbed MB at adsorption equilibrium was calculated using the equation:
where Q is the adsorbed MB at adsorption equilibrium (g MB g−1 carrier), C 0 is the initial MB concentration, C t is the MB concentration at time t, V is the solution volume, and W is the carrier weight.
The experimental procedure and mass transfer coefficient calculations were carried out according to Bai et al. (2010) and Akita and Yoshida (1973). 120-ml containers with 100 ml of 50 mg l−1 ammonium solutions and 5.0 g hydrogel carrier beads were used in these experiments. The samples were placed on a magnetic stirrer at a constant stirring speed of 500 rev min−1 while maintaining temperature at 30 °C. Diffusion of molecules in spherical-shaped beads with radius R can be appropriately modeled by the following relationship (3):
where D e (cm2 s−1) is the effective diffusion coefficient for the transport of the penetrant into the carriers and was monitored until there was no further change; C 0 is the initial ammonium concentration, C ∞ was determined from the ammonium concentration differential of 72 h, and R was the radius of the spherical hydrogel beads. So if we construct one figure with C t − C ∞ /C 0 − C ∞ versus t, then the slope could be determined as π2·D e ·R −2, and further D e can be calculated from k·R 2·π−2.
Analytical methods
NH4 +-N, NO2 −-N and NO3 −-N were measured by colorimetric methods according to Standard Methods (APHA 1995). pH measurement was done using a digital pH meter (PHS-25, Shanghai Leici Co. Ltd., China) while DO was measured by a digital DO meter (YSI, Model 55, USA). MLVSS concentration, used for the calculation of the biomass, was determined in accordance with the Standard Methods (APHA 1995). For SEM, the samples were first washed with phosphate buffer solution; then, fixed with 2.5 % glutaraldehyde at 4 °C overnight, washed with phosphate buffer solution twice; and then fixed in 1 % osmium tetroxide solution for 1 h, washed twice. The fixed cells were dehydrated by sequential immersion in increasing concentrations of alcohol (30, 50, 70, 80, 90, 95 and 100 %, v/v), washed twice in every concentration. Lastly, the dehydrated cells were immersed with acetonitrile (50, 70, 80, 90, 95, 100 %, v/v), 15 min every concentration, vacuumed, and sprayed carbon first, then sprayed gold. SEM observation was conducted using SEM model 5900LV (Hitachi, Japan).
Results and discussion
Specific surface area
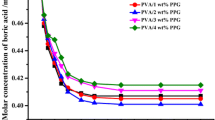
Relationships between the three kinds of biomass carrier hydrogel beads and MB concentration were developed as shown in the graphical representation of the Langmuir equation in Fig. 1. The values for Q max (mg MB g−1 carrier) were calculated from this graph. The maximum MB uptake capacities of PVA, PVA_SA, and PVA_SB hydrogel beads were 14.49, 23.26 and 26.32 mg MB g−1 carrier, respectively. The values of Q max obtained demonstrated that PVA_SA and PVA_SB beads had better MB adsorption capacity (about 1.64 and 1.85 times higher, respectively) than PVA beads. Q max represented adsorption saturation of the hydrogel beads and the specific surface area could be obtained by multiplying Q max by 2.45 (Hang and Brindley 1970). The corresponding specific surface areas of PVA, PVA_SA, and PVA_SB hydrogel beads were determined to be 35.26, 57.32 and 65.23 m2 g−1 carrier, respectively, illustrating that the addition of SA and SB resulted in increase in the specific surface areas by about 1.63 and 1.85 times compared to the conventional PVA hydrogel beads. This characteristic of PVA_SA and PVA_SB having higher specific surface areas was attributed to the pore-forming step, which allowed the hydrogel beads more porosity than conventional PVA hydrogel beads. Comparing PVA_SB hydrogel beads produced from our new technique with those of PVA_SA technique, the results demonstrated that PVA_SB beads had approximately 12 % higher surface area than PVA_SA beads. This characteristic makes PVA_SB hydrogel beads more attractive than PVA_SA and conventional PVA hydrogel beads by offering approximately 12–45 % higher space for the attachment and growth of microorganism.

Relationship of carrier weight with MB concentration in solution (closed square PVA, open circle PVA_SA, crotch PVA_SB)
Diffusion coefficient
Mass transfer resistance between the bulk liquid and the entrapped microorganisms in carriers is considered as a limiting factor for the entrapped microorganism to biodegrade pollutants. Therefore, determination of the diffusion coefficient of any carrier is of importance to ascertain pollutant removal performance by immobilization techniques. In this study, we determined the diffusion coefficient by measuring ammonium concentration in the solution with time. The c ∞ value was determined from the final steady ammonium concentration at the end of the experimental period. The D e value was determined from the slope of the linear relationship of −ln ((c t − c ∞ )/(c 0 − c ∞ )) and the diffusion time (Bai et al. 2010). The results are presented in Fig. 2.

Representative plots of the cumulative mass transfer capacity of ammonium into carriers with time (closed square PVA, open circle PVA_SA, open square PVA_SB)
The D e values of PVA, PVA_SA, and PVA_SB were calculated as 5.42 × 10−4, 7.0 × 10−4, and 7.49 × 10−4 cm2 s−1, respectively. The diffusion coefficients of PVA_SA and PVA_SB hydrogel beads were 29 and 38 % higher than the normal PVA carriers, respectively. This result of the two types of hydrogel carriers was consistent with that of the specific surface area results as previously described (Fig. 1). Similarly, the results also suggested that our newly developed PVA_SB hydrogel beads had a significantly higher diffusion coefficient than that of the PVA_SA hydrogel beads, making it desirable for application. This porous hydrogel carrier with high diffusion coefficient should allow more substrate to penetrate into the carrier at a faster rate and also more substrate to store within the carrier. At the same time, however, carriers with high diffusion coefficient are likely to leach microbial metabolites to the bulk solution from the carrier. The extent of transport of microbial metabolites into the bulk solution was not investigated in this study.
OUR and ammonium oxidation activity of nitrifying biomass
OUR is an important parameter to express microbial activity of biomass in wastewater treatment process. In our experiments, we not only examined the OUR of nitrifying biomass entrapped in the three types of PVA hydrogel beads but also the OUR of suspended nitrifying biomass for comparison purposes. The results are illustrated in Fig. 3. The OUR values of nitrifying biomass entrapped in PVA_SA and PVA_SB hydrogel beads were calculated as 16.62 and 19.53 mg O2 g MLVSS−1 h−1, respectively; which were approximately 26 and 49 % higher than that of nitrifying biomass entrapped in normal PVA hydrogel beads (OUR of about 13.15 mg O2 g MLVSS−1 h−1). As expected, the OUR of the suspended nitrifying biomass was higher than the entrapped nitrifying biomass and was determined to be 23.73 mg O2 g MLVSS−1 h−1. The reduced respiratory activity of entrapped nitrifying biomass is attributed to mass transfer resistance resulting from the immobilization process (Takei et al. 2011), and the effect of toxicity of boric acid on microorganisms (Zhang et al. 2007). Comparison of OUR of nitrifying biomass entrapped in PVA_SB with that of PVA_SA clearly shows that our newly developed PVA_SB technique enhanced respiratory activity of entrapped nitrifying biomass possibly due to higher surface area and porosity (also evident from the SEM photograph).

Comparison of oxygen uptake rate of the three kinds of hydrogel beads and un-entrapped free nitrifying biomass (VSS 0.221 g, 200 ml)
The results of ammonium-oxidizing activities of entrapped nitrifying biomass within the three PVA hydrogel beads demonstrated a trend similar to that of OUR experiments and are shown in Fig. 4. The ammonium-oxidizing activities of nitrifying biomass entrapped in PVA_SA and PVA_SB carriers were 9.57 and 10.59 mg N g MLVSS−1 h−1, respectively; while the activity of nitrifying biomass entrapped in the normal PVA carrier was only 7.40 mg N g MLVSS−1 h−1. Compared to that of the normal PVA hydrogel beads, the ammonium oxidation activities of nitrifying biomass entrapped in PVA_SA and PVA_SB hydrogel beads were improved by 29.4 and 43.2 %, respectively. The later results also suggested superior ammonium uptake of nitrifying biomass entrapped in our newly developed PVA_SB technique. Of course, the OUR rate of PVA_SB gel beads only accounted for about 82.4 % of that of the free biomass experiment. This implied that around 20 % activity would be decreased even with our newly developed PVA_SB gel beads.

Ammonium-oxidizing activity of nitrifying biomass entrapped in the three kinds of hydrogel bead carriers (VSS 0.221 g, 400 ml) (closed square PVA, open circle PVA_SA, open square PVA_SB)
Considering the results of OUR and ammonium oxidation activity together with results of specific surface area and diffusion coefficient, it is evident that the functioning of nitrifying biomass entrapped by the PVA_SB technique was the best followed by that of the PVA_SA technique. The improved OUR and ammonium oxidation activity of nitrifying biomass entrapped by PVA_SB technique could be attributed to the lower mass transfer resistance and higher specific surface area providing enhanced transportation of substrate.
SEM observation
SEM observation demonstrated that there were marked textural differences among the three kinds of PVA hydrogel beads, as shown in Fig. 5. The exterior surface of PVA and PVA_SA hydrogel beads illustrated a relatively smooth and even surface, while that of PVA_SB hyrogel beads had an uneven surface with lumps and folds cross-linked with each other (Fig. 5a–c, 10,000×). Dave and Madamwar (2006) found that addition of alginate could prevent agglomeration between hydrogel beads, and allow beads with higher hydrogel mechanical strength, which could be the reason for smooth surface of PVA_SA hydrogel beads. Since the surface of PVA hydrogel beads was also smooth and even, it is unclear if there were other factors impacting the smoothness of the beads. The observed specific protuberance could explain why the PVA_SB hydrogel beads possessed the highest specific surface area.

SEM photographs of PVA hydrogel bead carriers. a Outside the PVA carrier, scale bar 10 μm; b outside the PVA_SA carrier, scale bar 10 μm; c outside the PVA_SB carrier, scale bar 10 μm, d inside the PVA carrier, scale bar 1 mm; e inside the PVA_SA carrier, scale bar 1 mm; f inside the PVA_SB carrier, scale bar 1 mm, g inside the PVA carrier, scale bar 20 μm; h inside the PVA_SA carrier, scale bar 20 μm; and i inside the PVA_SB carrier, scale bar 20 μm
Scanning electron micrographs of the cleaved hydrogel beads in Fig. 5d–f (100×) revealed striking dissimilarity in the interior of the three kinds of hydrogel beads. The variation could be clearly observed in the enhanced magnification micrographs of Fig. 5g–i (5,000×).
The interior structure of a normal PVA hydrogel bead appeared to be a relatively dense pinhole-like structure extending uniformly over the entire bead (Fig. 5d) whereas that of PVA_SA and PVA_SB beads manifested a quasi-foamy layered appearance (Fig. 5e, f). The PVA_SA bead seemed to have 3 layers with the outer layer composed of 0.06–0.1 mm diameter circular to elliptical pores (Fig. 5e).The second layer appeared to have relatively larger (0.3–0.5 mm diameter) pores. The core of the bead formed the central layer and appeared to be more dense, resembling somewhat the interior of the PVA bead. The interior of the PVA_SB bead (Fig. 5f) appeared to have an outer layer fairly similar to that of PVA_SA bead (Fig. 5e) and a central part that showed dense sags and folds with a lesser extent of 0.03–0.5 mm diameter pores.
Higher magnification micrographs of the dense areas of the interior of these three kinds of hydrogel beads in Fig. 5g–i (5,000×) illustrated structural formation of the beads. These micrographs suggested that beads prepared by normal PVA and PVA_SA techniques shared almost similar honeybee web-like microstructure. However, the micro-configuration of hydrogel beads prepared by our newly developed PVA_SB technique was completely different compared with the other two kinds of hydrogel beads. The PVA_SB hydrogel beads demonstrated dendritic-like structures that were observed to be cross-linked with each other and formed larger gaps having diameter of approximately 10–40 μm (Fig. 5i). The formation of this kind of structure could be attributed to the addition of sodium bicarbonate, which could change the inner structures by micro CO2 gas bubbles produced in the bead preparation process. It was, therefore, determined that hydrogel beads prepared by PVA_SB technique could provide larger space for microorganism attachment and growth inside these beads, lower mass transfer resistance, increased OUR, and result in improved pollutant removal later.
Conclusion
In conclusion, the addition of sodium bicarbonate proved to be beneficial for improving the specific surface area of PVA_SB hydrogel beads (65.23 m2 g−1 carrier). Moreover, it reduced the mass transfer resistance by increasing its diffusion coefficient to 7.49 × 10−4 cm2 s−1, which was 38 % higher compared to those of normal PVA hydrogel beads. The OUR and nitrification rates of PVA_SB entrapped nitrifying biomass were measured as 19.53 mg O2 g MLVSS−1 h−1 and 10.59 mg N g MLVSS−1 h−1, respectively; which were 49 and 43 % higher than those of normal PVA hydrogel beads. SEM observation demonstrated that there existed cross-linked protuberances on the outer surface of PVA_SB hydrogel beads, which might be the main reason for the relatively higher specific surface area. The observed loose micro-structures of the inner parts of these beads were considered as an important explanation for mass transfer resistance reduction and microbial activity improvement.
References
Akita K, Yoshida F (1973) Gas holdup and volumetric mass transfer coefficient in bubble columns. Effects of liquid properties. Ind Eng Chem Process Des Dev 12:76–80
APHA (American Public Health Association) (1995) Standard methods for the examination of water and wastewater, 19th edn. APHA, Washington
Ariga O, Takagi H, Nishizawa H, Sano Y (1987) Immobilization of microorganisms with PVA hardened by iterative freezing and thawing. J Ferment Technol 65:651–658
Bai X, Ye Z, Li Y, Zhou L, Yang L (2010) Preparation of crosslinked macroporous PVA foam carrier for immobilization of microorganisms. Proc Biochem 45:60–66
Bezbradica D, Obradovic B, Leskosek-Cukalovic I, Bugarski B, Nedovic V (2007) Immobilization of yeast cells in PVA particles for beer fermentation. Proc Biochem 42:1348–1351
Chirizzi D, Guascito MR, Malitesta C, Mazzotta E (2011) Electrochemical and spectroscopic characterization of glucose oxidase immobilized in polyvinyl alcohol and applications in glucose detection. In: Neri G et al (eds) Sensors and microsystem (AISEM 2010 proceedings). Springer, Berlin, pp 339–343
Chou WL, Wang CT, Hsu CW, Huang KY, Liu TC (2010) Removal of total organic carbon from aqueous solution containing polyvinyl alcohol by electrocoagulation technology. Desalination 259:103–110
Dave R, Madamwar D (2006) Esterification in organic solvents by lipase immobilized in polymer of PVA-alginate-boric acid. Proc Biochem 41:951–955
El-Naas MH, Al-Muhtaseb SA, Makhlouf S (2009) Biodegradation of phenol by Pseudomonas putida immobilized in polyvinyl alcohol (PVA) gel. J Hazard Mater 164:720–725
Hang PT, Brindley GW (1970) Methylene blue absorption by clay minerals. Determination of surface areas and cation exchange capacities (clay-organic studies XVIII). Clays Clay Miner 18:203–212
Hashimoto S, Furukawa K (1987) Immobilization of activated sludge by PVA-boric acid method. Biotechnol Bioeng 30:52–59
Khan R, Banerjee UC (2010) Decolorization of azo dyes by immobilized bacteria. In: Hatice AE (ed) Biodegradation of Azo Dyes. Springer, Berlin, pp 73–84
Khoo KM, Ting YP (2001) Biosorption of gold by immobilized fungal biomass. Biochem Eng J 8:51–59
Lei S, Wang K, Lu X, Pan S (2011) Immobilization of Naringinase on PVA-calcium alginate gel. Food Sci 32:138–143
Puppi D, Piras AM, Detta N, Ylikauppila H, Nikkola L, Ashammakhi N, Chiellini F, Chiellini E (2011) Poly (vinyl alcohol)-based electrospun meshes as potential candidate scaffolds in regenerative medicine. J Bioact Compat Pol 20:20–34
Qiao S, Matsumoto N, Shinohara T, Nishiyama T, Fujii T, Bhatti Z, Furukawa K (2010) High-rate partial nitrification performance of high ammonium containing wastewater under low temperatures. Bioresour Technol 101:111–117
Quan LM, Khanh DP, Hira D, Fujii T, Furukawa K (2011) Reject water treatment by improvement of whole cell anammox entrapment using polyvinyl alcohol/alginate gel. Biodegradation 22:1155–1167
Ricciardi R, Auriemma F, De Rosa C, Lauprêtre F (2004) X-ray diffraction analysis of poly (vinyl alcohol) hydrogels, obtained by freezing and thawing techniques. Macromolecules 37:1921–1927
Sandeman SR, Gun’Ko VM, Bakalinska OM, Howell CA, Zheng Y, Kartel MT, Phillips GJ, Mikhalovsky SV (2011) Adsorption of anionic and cationic dyes by activated carbons, PVA hydrogels and PVA/AC composite. J Colloid Interface Sci 358:582–592
Surmacz-Gorska J, Gernaey K, Demuynck C, Vanrolleghem P, Verstraete W (1996) Nitrification monitoring in activated sludge by oxygen uptake rate (OUR) measurements. Water Res 30:1228–1236
Takei T, Ikeda K, Ijima H, Kawakami K (2011) Fabrication of poly (vinyl alcohol) hydrogel beads crosslinked using sodium sulfate for microorganism immobilization. Proc Biochem 46:566–571
Tripathi S, Mehrotra GK, Dutta PK (2009) Physicochemical and bioactivity of cross-linked chitosan-PVA film for food packaging applications. Int J Biol Macromol 45:372–376
Wang S, Zhu ZH, Coomes A, Haghseresht F, Lu GQ (2005) The physical and surface chemical characteristics of activated carbons and the adsorption of methylene blue from wastewater. J Colloid Interface Sci 384:440–446
Wang YJ, Yang XJ, Li HY, Tu W (2006) Immobilization of Acidithiobacillus ferrooxidans with complex of PVA and sodium alginate. Polym Degrad Stab 91:2408–2414
Wang YJ, Yang XJ, Tu W, Li HY (2007) High-rate ferrous iron oxidation by immobilized Acidithiobacillus ferrooxidans with complex of PVA and sodium alginate. J Microbiol Methods 68:212–217
Wu KYA, Wisecarver KD (1992) Cell immobilization using PVA crosslinked with boric acid. Biotechnol Bioeng 39:447–449
Yu Y, Zhuang YY, Wang ZH (2001) Adsorption of water-soluble dye onto functionalized resin. J Colloid Interface Sci 242:288–293
Zhang LS, Wu WZ, Wang JL (2007) Immobilization of activated sludge using improved polyvinyl alcohol (PVA) gel. J Environ Sci 19:1293–1297
Acknowledgments
This work was supported by the Natural Science Foundation of China (Nos. 21377014, 51008045), the Fundamental Research Funds for the Central Universities (No. DUT12LK20) and the Dalian Municipal Science and Technology Projects (No. 2012E11SF067) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Qiao, S., Duan, X., Zhou, J. et al. Enhanced efficacy of nitrifying biomass by modified PVA_SB entrapment technique. World J Microbiol Biotechnol 30, 1985–1992 (2014). https://doi.org/10.1007/s11274-014-1621-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-014-1621-4