
Abstract
In the present study nine promising primer sets, targeting Archaea and methanogenic Archaea in particular, were evaluated in silico, in vitro and in situ concerning specificity, accuracy and applicability in end-point (ep-) and especially quantitative (q-)PCR research. The main goal was to adapt and evaluate already adapted primer sets, which were partially designed in combination with TaqMan probes, in substantially cheaper SYBR Green-based qPCR applications. An initial 16S rRNA gene bank-based in silico evaluation revealed high coverage potentials for all primers within targeted groups, ranging from 71 to 90 %, except the Methanosaeta specific set showing a low potential of 37 %. Mentionable cross-reacting potentials could be detected for the Methanothermobacter, Methanomicrobiales and Methanoculleus sets. The in vitro evaluation with selected reference organisms revealed a specific behavior for most primer sets, while the Methanosarcina and Methanothermobacter sets showed most problematic cross-reactions in epPCR application. We were able to show that primers for detecting the total archaeal community, methanogenic orders Methanosarcinales, Methanobacteriales, Methanococcales and the genus Methanoculleus performed in a highly specific way and allowed an accurate quantification of targeted organisms without the use of expensive TaqMan probes. However, primer pairs designed for detecting Methanomicrobiales, Methanothermobacter, Methanosarcina and Methanosaeta are not suitable for SYBR Green applications. The reliability of in situ quantifications was assessed for a typical methanogenic community, derived from a thermophilic fermenter, and confirmed via denaturing gradient gel band quantification and sequencing. Thereby, we revealed high abundances of methanogenic Archaea, mainly comprising Methanoculleus and Methanosarcinales, while Methanobacteriales only formed a minor fraction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The detection and quantification via quantitative PCR (qPCR) is a promising tool to reveal conditions and alterations within microbial communities. For a huge variety of microbial groups target-oriented primers have been developed (Franke-Whittle et al. 2009; Yu et al. 2005). Nevertheless, primers can be regularly seen (re-)evaluated or proved as non-specific or not universal (Baker et al. 2003). By default, primers are designed and evaluated in silico, by aligning them to a representative quantity of sequenced target organisms in online databases (Abd-Elsalam 2003). The primer design process is a first approach but does not represent an overall reliable evaluation, since these databases are neither immune to errors nor complete and cannot simulate 100 % realistic reaction behavior of a PCR. Neither an in vitro evaluation of primers via specificity studies (pure-culture DNA from known reference organisms) nor in situ (applying complex DNA from various environments) is encompassing. However, this is the only way to gather sufficient information about whether a certain primer pair can be applied to a particular habitat. Otherwise, non-specific primers will lead to significant altered results, which may not represent the reality within the community.
Today’s most established detection systems in qPCR applications are the amplicon quantification with the dsDNA-binding SYBR Green dyes and the more specific approach via a third detection probe (e.g., TaqMan probes) (Zhang and Fang 2006). However, these latter detection systems are substantially more expensive than SYBR Green-based systems. Furthermore, SYBR Green reaction mixes can in principle be applied to every desired primer-pair, while probes can only be used in combination with a certain primer pair.
The aim of the present study was to evaluate the specificity of a representative repertory of nine commonly used primer pairs for detecting and quantifying the most relevant members within methanogenic communities in end-point (ep)PCR and quantitative (q)PCR and their adaptability to SYBR Green-based qPCR applications. Although primers targeting functional genes are available, most of these primers aim at higher-ranking taxonomic units, like mcrA gene specific primers do. The mcrA gene encodes for a peptide within the methyl coenzyme-M reductase, the enzyme complex which catalyzes the coenzyme-bound methyl reduction and subsequent release of methane, which is thought to be common in all methanogens (Luton et al. 2002). We focused on 16S rRNA-coding genes and corresponding primer sets in order to discriminate between closely-related methanogenic taxa. All primers were checked in silico, in vitro and in situ to assess their suitability in studies dealing with complex methanogenic environments. The reference organisms for the in vitro evaluation were chosen according to results of former studies (Lins et al. 2010; Malin and Illmer 2008; Wagner et al. 2011), under the terms of a potential occurrence within diverse methanogenic habitats and with regard to cover a wide phylogenetic range. The main goal of this study is not to devalue other researchers’ work. However, considering that previously published papers are supposed to present information that is usable without further evaluation and that previously published primers and adaption methods are seldom questioned, the aim of this paper—besides the strict evaluation of methanogenic primers—is to evaluate the praxis of primer adaption from TaqMan to SYBR Green qPCR assays and to question insufficiently evaluated primer publications.
Materials and methods
To differentiate between a quantitative real-time (q)PCR and a conventional PCR, and in terms of confirmability, we will refer to it as end-point (ep)PCR.
In silico evaluation of primers—phylogenetic coverage potential
The selection of the primer pairs was based on extensive preliminary paper research, choosing the most promising and up-to-date primers developed for investigating methanogenic habitats. Table 1 lists the primers to be evaluated in this study and their specifications. To reveal potential self-annealing or dimer formation, primers were at first checked with AutoDimer software (version 1.0) (Vallone and Butler 2004). Following that, primers were analyzed in silico with the TestPrime 1.0 application (Klindworth et al. 2013) using the SILVA SSU r114 RefNR database (Quast et al. 2013), available at the SILVA homepage (http://www.arb-silva.de). This tool makes it possible to align the sequences with verified prokaryotic and eukaryotic sequences in the SILVA comprehensive ribosomal RNA databases and evaluates their performance by simulating a virtual polymerase chain reaction (PCR) (Pruesse et al. 2007). In line with the developer’s instructions, and to simulate most realistic PCR behavior, stringency parameters were set as follows: maximum number of mismatches = “1 mismatch”, length of 0-mismatch zone at 3’ end = “5 bases”; with exception of the Archaea- and Methanomicrobiales-specific primer sets, where stringency conditions had to be set to “0 mismatch” to receive reliable results (Table 2). We evaluated the coverage potential within most important methanogenic groups and in particular of those groups that were most important with regards to our research objects (e.g., anaerobic fermenters and soil). The database dimensions of evaluated groups were: 10581 (Arc, Archaea), 879 (MSL, Methanosarcinales), 332 (Mesae, Methanosaeta), 379 (Methanosarcinaceae), 188 (Mesa, Methanosarcina), 23 (Methanomethylovorans), 559 (MMB, Methanomicrobiales), 136 (Mecu, Methanoculleus), 73 (Methanospirillum), 592 (MBT, Methanobacteriales), 115 (Methanobacterium), 29 (Meth, Methanothermobacter) and 61 sequences in case of MCC (Methanococcales).
In vitro evaluation of primers—specificity and accuracy
The term specificity addresses the extent to which a certain primer pair in a certain assay is able to detect its targeted phylogenetic group only, while the term accuracy addresses the ability of a certain primer pair/assay to detect the exact number of individuals given.
To evaluate the in vitro specificity of chosen primers, mesophilic and thermophilic reference organisms were selected, covering the major groups of methanogenic Archaea and reflecting typical anaerobic habitats: Methanosarcina acetivorans (Meac, DSM No. 2834), Methanosarcina barkeri (Meba, DSM No. 800), Methanosarcina thermophila (Mela, DSM No. 1825), Methanomethylovorans thermophila (Meme, DSM No. 17232) and Methanosaeta concilii (Meco, DSM No. 2139) from the Methanosarcinales (MSL); Methanoculleus bourgensis (Mebo, DSM No. 3045), Methanoculleus thermophilus (Meph, DSM No. 2373) and Methanospirillum hungatei (Mehu, DSM No. 864) from the Methanomicrobiales (MMB); Methanobacterium thermaggregans (Meag, DSM No. 3266), Methanobacterium formicicum (Mefo, DSM No. 1535), Methanothermobacter thermautotrophicus (Meth, DSM No.3720) and Methanothermobacter wolfeii (Mewo, DSM No. 2970) from the Methanobacteriales (MBT); and Methanococcus voltae (Mevo, DSM No. 1537) from the Methanococcales (MCC). As out-groups anaerobic as well as aerobic Gram-positive bacteria: Listeria monocytogenes (Limo, DSM No. 20600) and Coprothermobacter proteolyticus (Copr, DSM No. 5265), Gram–negative bacteria: Methylobacter tundripaludum (Metu, DSM No. 17260), Escherichia coli (Esco, DSM No. 5347) and Thermotogae lettingae (Thle, DSM No. 14385), and fungal representatives: Geotrichum klebahnii (Gekl) were chosen. All prokaryotes were obtained from the DSMZ (German Collection of Microorganisms and Cell Cultures) as active growing cultures or DNA in case of no or low growth yields, while the yeast was isolated out of compost (Illmer et al. 2007).
The active growing cultures were cultivated as recommended by DSMZ and 700 μL of each culture were applied for DNA extraction with the NucleoSpin Soil DNA extraction kit (Macherey–Nagel). Quality and quantity of all DNA species were checked via agarose gel electrophoresis, spectrophotometry on a NanoDrop 2000c (Thermo Scientific) as well as with PicoGreen-based fluorometric detection (Quant-iT) on a Zenyth 3100 multimode fluorometer (Anthos) to obtain highly precise DNA quantities. Afterwards the DNA extracts were diluted in DNase/RNase-free distilled water from gibco life technologies (A. d., aqua destillata) to final concentrations of 1.0 and 0.2 ng μL−1 respectively before being applied in ep- and qPCR applications for evaluating specificity and sensitivity. To gain comparable information in the assays, we added equivalent amounts of genomic DNA per PCR approach. Preliminary tests showed that 0.2 ng μL−1 DNA concentrations were sufficient for qPCR-based evaluation approaches, while, due to the lower sensitivity regarding epPCR/GE-based approaches, DNA concentrations had to be increased to 1.0 ng μL−1 in this case.
Original and modified PCR conditions are stated in Table 3. The melting temperatures were recalculated with OligoAnalyzer software (version 3.1; Integrated DNA Technologies [http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer/default.aspx]) and corresponded well to the original specifications given for qPCR applications, but not concerning epPCRs. In the case of epPCR applications, dNTPs were set to 0.2 mM, while primer concentrations were set to 0.2 (Arc, Mesa, Mesae, Meth and Mecu) or 0.4 (MSL, MCC, MBT and MMB) μM. 0.625 units of DNA polymerase (BioTherm) were added per epPCR approach. Concerning qPCR, we increased the MgCl2 concentration to 0.5 mM, while primer concentrations were in most cases decreased to 0.15 (Mesae), 0.2 (Mesa and Meth), 0.25 (Arc, MCC and MMB) or 0.3 (MSL, MBT and Mecu) μM.
Quantitative PCRs were performed with the SensiMix SYBR No-ROX kit (Bioline) on a Corbett Life Science (Qiagen) Rotor-Gene 6000 system. All PCR reactions and approaches were performed at least in duplicates, with no template controls (NTCs), positive and negative controls. One μL of template-DNA was added per epPCR, while two μL were added per qPCR approach. The final volumes of ep- and qPCR approaches were 25 and 20 μL respectively. For quantification and efficiency calculation we used diluted standards and plotted the CT (cycle threshold) values against the log of given templates to gain standard graphs (Bustin et al. 2009). As standards we used purified PCR products of known concentrations. Therefore we applied pure DNA from a corresponding reference organism for each primer pair (Meac for Arc primer, Meac for MSL primer, Meba for Mesa primer, Meco for Mesae primer, Mehu for MMB primer, Mebo for Mecu Primer, Meth for Meth primer, Mewo for MBT primer and Mevo for MCC primer) in conventional epPCRs, purified and quantified the amplicons as described above. Subsequently, the copy numbers were calculated, based on the measured DNA amount (ng) and on the verified amplicon length (bp) as described in Yun et al. (2006). Stock solutions with 1E+09 copies μL−1 were prepared, aliquoted and stored frozen. Fresh batches were prepared shortly before use. For calibration curves, dilutions from 1E+08 to 1E+02 were applied in duplicates (dynamic range). The efficiency calculation itself was performed by the Rotor-Gene software. From all epPCR approaches and representative spot tests of qPCR, runs were checked via gel electrophoresis. All qPCR runs were checked with melt curve analysis, with temperature intervals of 0.2 °C, subsequent to amplification (provided as supplement 02, “In vitro qPCR”).
To draw conclusions regarding specificity, sensitivity and accuracy of the adapted qPCR applications with the selected primer sets, copy numbers of 16S rRNA genes were calculated based on added reference DNA amounts (Einen et al. 2008) and compared with measured values (Figs. 1, 2). Copy numbers per genome, of each reference organism, were obtained from the rrndb, the Ribosomal RNA Operon Copy Number Database (http://rrndb.mmg.msu.edu), while for non-listed species the averaged copy number of the corresponding genus was used. Organismic genome sizes could be determined for Meac (Galagan et al. 2002), Meba (Zhu et al. 2012), Mela (Diaz-Perez et al. 1997), Meco (Zhu et al. 2012), Mebo (Maus et al. 2012), Mehu (Anderson et al. 2009), Meth (Fogel et al. 1999), Mewo (Fogel et al. 1999), Mevo (Fogel et al. 1999), Limo (Fogel et al. 1999), Copr (Koonin and Wolf 2008), Metu (Dam et al. 2012), Esco (Fogel et al. 1999), Thle (Blumer-Schuette et al. 2008) and Gekl (Gregory et al. 2006), while for Meme, Meph, Meag and Mefo averaged genome numbers, characteristic for their taxon, were chosen. For more species specific information see the additional table entitled “Reference Organisms” (supplement 01).
In situ measurements and confirmation of specificity
To evaluate the performance of most specific primer sets in best adapted ep- as well as qPCR applications, an in situ evaluation was performed with samples of a typical methanogenic habitat, a thermophilic anaerobic digester. The investigated samples were obtained from the control approach of the fermenter sludge experiment described in Lins et al. (2012), where diluted fermenter sludge was incubated in a modified DSMZ 119 medium with 150 mM acetate for 77 days in triplicates. During the experiment samples were collected at days 0, 10, 21, 31, 42, 56 and 77. DNA extraction and checks of fermenter sludge (FS) samples as well as qPCRs were carried out as described above and listed in Table 3. Temperature intervals of the melt curve analyses were set to 0.5 °C. Standard graphs and melt curves are provided as supplementary material (supplement 03, “In situ qPCR”). Following that, the overall Archaeal community was investigated using denaturing gradient gel analysis (DGGE), and a representative collection of DNA bands was excised after separation. For the DGGE analyses a GC clamp (Muyzer et al. 1993) was attached to the 5′-end of the Arc forward primer and an epPCR was conducted with the conditions described above and listed in Table 3. The DGGE protocol was altered based on the work of Sekiguchi et al. (2001). For the separation an INGENYphorU electrophoresis system was used (60 °C, 100 V for 16 h). The acrylamide concentration in the gel was between 7 and 8 %, while urea and formamide concentrations were between 35 and 70 %. After separation, DNA bands were stained with silver nitrate. Distinct DNA bands were quantified relatively. For this, densitometric curves of each DGGE lane were readout with ImageJ software (available at: http://rsb.info.nih.gov/ij/), after background subtraction. Afterwards, an averaged threshold was determined, and peak areas were integrated and set into relation to the sum of peak areas per lane. To obtain qualitative data, products were separated in a new DGGE and stained with SYBR Gold Nucleic Acid Gel Stain (Invitrogen). Equivalent, previously quantified bands were excised under UV light, suspended in A. d. and used as template in a new PCR with the Arc primer pair, but without GC clamp to obtain better sequencing results. Positive PCR products were purified with NucleoSpin Extract II (Macherey–Nagel) and sequenced by Eurofins MWG Operon (Ebersberg, Germany). Passed sequences were processed and checked concerning quality with CLC DNA Workbench 5.6.1 (CLC bio) and aligned via NCBI Blast tool (http://blast.ncbi.nlm.nih.gov/).
Finally, the quantitative data gained via qPCR were compared to the quantitative–qualitative DGGE and sequencing results to reason the reliability of SYBR Green-based qPCR adapted in this study.
Results
All primer pairs that were evaluated in the study regarding specificity, accuracy and applicability in end-point and, most importantly, in SYBR Green-based quantitative PCR applications are shown in Table 1. This includes the summary of the evaluation, giving a recommendation whether a SYBR Green-based quantification is advisable or not.
In silico evaluation of primers—phylogenetic coverage potential
Table 2 summarizes the coverage potential of chosen primers within selected methanogenic groups. While the total coverage of the Arc primers is quite low (70.5 %), the coverage within methanogenic Euryarchaeota is substantially higher, ranging from about 80 to 100 %. Furthermore, no potential cross-reactivities regarding bacteria or Eukaryota could be determined. The MSL set had a potential of 76 % to detect members within Methanosarcinales, while members of the genus Methanosaeta are only covered to 68 % compared to 89 % concerning Methanosarcina. A slight cross-reacting potential of 0.5 % is given for Methanolinea (MMB). The Mesa set showed a high coverage of 82.9 % within the target group and only negligible cross-reactions of 0.2 % in the C19A group (MMB) and concerning ANME-3 and Methanolobus (Methanosarcinaceae, MSL). The lowest affinity to its target group could be confirmed for the Mesae primers, detecting only 37 % of available sequences, with a negligible cross-reaction potential of 0.3 % within uncultured Methanosarcinaceae (MSL). A potential of 75 % for specific coverage is given with the MCC set, showing only slight false-positive detection potential for pCIRA-13 (Euryarchaeota). The MBT primer set performed in a highly specific way, covering 90 % of all Methanobacteriales, 88 % of Methanobacterium and 100 % of Methanothermobacter. However, a negligible cross-reaction potential concerning uncultured Halobacteria with 0.1 % and Thermoplasmata with 0.2 % (both Euryarchaeota) was revealed. Even though the Meth set showed highly specific coverage (83 %) within Methanothermobacter, a proportionally high percentage of 8 % within the closely related Methanobacterium species was also covered. Basically, the MMB set showed a moderate coverage of 73 % within Methanomicrobiales, 88 % within Methanoculleus and 79 % within Methanospirillum, although detecting members within Methanosarcina (1.4 %), Methanosaeta (2.3 %) and Methanocalculus, all belonging to the MSL order. The Mecu set highlighted a high coverage of 89 % within the targeted group. Slight cross-reactions could be detected in closely related groups of Methanospirillum (1.5 %), Methanolinea, Methanosphaerula and Methanofolis (all MMB) and members within the MSL cluster (0.4 %).
In vitro evaluation of primers—specificity and accuracy
As summarized in Table 3, additions and massive alterations compared to the original specifications were necessary to obtain reliable and specific measurements. Although we observed high efficiencies with initially applying 2-step qPCR variants, most NTCs were false-positive and primer dimers were detectable. Therefore, we decided to discard 2-step assays in general and changed to 3-step variants for all primer pairs. To get rid of non-specific products and primer dimers in ep- as well as qPCRs as far as possible, drastically lower primer concentrations, higher annealing temperatures and shorter elongation times had to be applied. Although amplification efficiencies could not be increased or kept constant in all cases, clearly noticeable for Mesae and MCC primers (Table 3), corresponding qPCR conditions were chosen as the best compromise between specificity and efficiency.
All primer pairs could detect their corresponding reference organisms. Primer pairs Arc, MSL, Mesae, MBT and Mecu showed highly specific results and are therefore suitable for further epPCR-based qualitative research on methanogenic communities (e.g., PCR-DGGE-based investigations) for assessing microbial diversity and for sequencing purposes. Primer pairs Mesa, MCC, Meth and MMB revealed mentionable non-specificities, with most problematic cross reactions of the Mesa set detecting the closely related Methanomethylovorans thermophila (MSL) and with the Meth set detecting the closely related Methanobacterium thermaggregans (MBT).
Compared to the results of end-point amplifications, quantitative detection and analysis via SYBR Green-based real-time PCR revealed considerably higher non-specificities within certain primer sets, although highlighting primer sets Arc, MSL, MBT and MCC as well as Mecu to be valuable tools for specific and accurate quantification of targeted methanogens. To gain information about the sensitivity and accuracy of the primer sets used in the adapted qPCR applications, the added 16S rRNA gene copy numbers, calculated according to actually applied DNA amounts, were compared with the measured gene copy numbers in vitro. As can be seen in Figs. 1 and 2, the actually measured copy numbers of Archaea, Methanosarcinales, Methanobacteriales and Methanococcales as well as Methanoculleus cluster are close to the calculated, added number of copies, while non-target organisms are either not amplified (MSL) or in case of false-positives the quantification results are about 1–3 (MBT), 2.5–4.5 (Mecu) and 3.5–4.5 (MCC) orders of magnitude lower and alter the quantification result to a negligible or slight extent. In case of Archaea we observed higher non-specific reactions concerning bacterial and fungal representatives, although the averaged distance between targeted Archaea and false-positives was still 2 orders of magnitude, altering the Archaeal quantification. Although, due to the limited quantitative impact on the quantification results their appliance is acceptable, if a careful pre- and post-analytical evaluation of the results is carried out. Further information is provided in supplement 04 (“In vitro qPCR—selected primers).

Specificity and accuracy of Archaea (Arc) and methanogenic order specific primers (MSL, MCC, MBT and MMB) were checked in vitro in adapted SYBR Green-based qPCR applications. Normalized amounts of DNA from reference organisms were added per qPCR approach and corresponding 16S rRNA gene copy numbers were calculated [cop (qPCR)]. These calculations are compared with the copy numbers actually measured, obtained from the calibrated qPCR runs with the corresponding primer sets. Encircled areas visualize the quantification of order specific primers within targeted groups. It is clear that primer pairs Arc, MSL, MCC and MBT show no or slight unspecific amplification behavior, by detecting false-positives in several orders of magnitude less than targeted groups

Specificity and accuracy of methanogenic genus specific primers (Mesa, Mesae, Meth and Mecu) were checked in vitro in adapted SYBR Green-based qPCR applications. Normalized amounts of DNA from reference organisms were added per qPCR approach and corresponding 16S rRNA gene copy numbers were calculated [cop (qPCR)]. These calculations are compared with the copy numbers actually measured, obtained from the calibrated qPCR runs with the corresponding primer sets. Encircled areas visualize the quantification of genus specific primers within targeted groups. It is clear that only primer pair Mecu showed a slight unspecific amplification behavior, by detecting false-positives in several orders of magnitude less than targeted groups
Even though the MMB set showed a highly accurate quantification of target organisms, we were able to observe extensive cross-reactions, altering quantitative results more considerably (Fig. 1). The quantification values of primer pair Mesa showed an extremely broad distribution pattern, detecting target organisms added in concentrations of about 1E+05 copies per reaction, in quantities of 1E+03 to 1E+08, therefore making their application in a SYBR Green-based qPCR not reliable (Fig. 2). Furthermore, the high quantification values of many non-target reference organisms excludes the primer pair from SYBR Green quantification. In case of the Mesae set, the low affinity to targeted Methanosaeta could be proved and moreover extensive cross-reactivities (Fig. 2), also excluding this primer pair. A different reaction behavior was detectable for the Meth set. Even though reference M. wolfeii was detected to almost 100 %, reference M. thermautotrophicus was detected in 2 orders of magnitude lower quantities (Fig. 2). In connection with the constant detection of all applied references in mentionable quantities, most likely due to a universal amplification behavior of the primers, a reliable quantification with the Meth set could not be proved.
In situ measurements and confirmation of specificity
Based on the results from the in silico and in vitro evaluation of all primer sets, the most specific primer pairs and the most reliable SYBR Green-based qPCR adaptions were applied for investigating an anaerobic digester community.
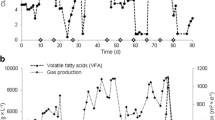
Highest abundances in qPCR were obtained with Arc specific primers, revealing values between 6E+07 and 5E+09 Archaea mL−1 during the fermenter sludge (FS) experiment, while the Mecu set revealed Methanoculleus, with 2E+07 to 2E+09 organisms mL−1 FS, to be the most abundant methanogenic genus (Fig. 3). As second-most abundant methanogens the Methanosarcinales could be addressed, with the MSL set detecting 8E+06 to 1E+08 organisms mL−1 FS, becoming the most abundant methanogens in the end of the cultivation (Fig. 3). The MBT primer pair illustrated significantly (p < 0.001) lower abundances of Methanobacteriales compared to Arc, Mecu and MSL after 10 days of incubation, which were between 6E+05 and 8E+06 mL−1 FS (Fig. 3). No members of the Methanococcales were detected.

The quantitative development of the total archaeal community (Arc)—representative for the total methanogenic community in the anaerobic batch experiment with fermenter sludge—and of the orders Methanosarcinales (MSL) and Methanobacteriales (MBT), as well as the genus Methanoculleus (Mecu) is displayed in course of 77 days of incubation in an acetate containing medium
Based on the semi-quantitative DGGE band determination and combined sequencing results, we were able to show that all bands amplified with the Arc primer set were of Archaeal and methanogenic origin, emphasizing the appropriateness of the Arc set for quantifying methanogens in fermenter sludge. Furthermore, we could show that members of Methanoculleus (MMB order) form the most abundant genus. Related bands contributed to approximately 100 % of total bands at the beginning of the incubation and decreased to about 50 % at the end (Fig. 4). Bands affiliated to members of the Methanosarcinales (basically Methanosarcina, with negligible Methanomethylovorans contribution) raised from about 0 to 50 % during the in situ experiment (Fig. 4). No members of Methanobacteriales or Methanococcales were found, due respectively to their low abundance and absence of corresponding representatives. The relative qPCR values of MSL and Mecu, in percentage of the total Archaea, affirm the DGGE/sequencing results. The high congruence is most evident in case of the MSL increase during the incubation experiment. The trend of Methanoculleus is not as clear, although highlighting that Mecu form the dominant methanogenic group in the fermenter sludge experiment. While members of MBT were detected during qPCR, together with unidentified organisms contributing to the total archaeal community and the difference that is not represented in DGGE/sequencing data. However, the main message that all detected Archaea are methanogens and the two dominating groups are Methanoculleus and Methanosarcinales could be shown by this unconventional approach.

The absolute percentage of the dominant methanogens, the genus Methanoculleus (Mecu) and the order Methanosarcinales (MSL), within the total archaeal community during the anaerobic fermenter sludge experiment, was measured via qPCR and compared to the semi-quantitative values obtained via DGGE peak area quantification and band affiliation via sequencing, in order to emphasize the reliability of the selected primers for in situ measurements on fermenter sludge communities
Discussion
This study does not claim to present exhaustive evaluated primers, because this is not entirely feasible considering the huge variety of parameters and their combination possibilities. Primers are created on basis of described and sequenced organisms. This may lead to an underestimation of the actual abundance of a certain group due to a low or no coverage within unknown and genomic differing members within a phylogenetic group (Murrell and Jetten 2009). Complete coverage of related microorganisms with this technique is therefore unlikely. Nevertheless, the assessment and the quantification of microorganisms based on 16S rRNA genes is still a valuable and common tool (Franke-Whittle et al. 2009; Skillman et al. 2004; Tatara et al. 2008; Yu et al. 2006). Our goal was to transfer a representative set of primers for detecting methanogens from the expensive TaqMan system, where the costs for the additional probe is about ten times higher than for a standard primer, to the more economic SYBR Green system.
After the in silico evaluation, primer pairs Mesae (due to the considerable low target-specific coverage), Meth (due its significant high coverage within a closely related group), MMB and Mecu (due to their slight, but broad physiological coverage within other methanogenic groups) were especially checked before application. The in vitro evaluation of all primers with reference organisms revealed a more distinct non-specific behavior regarding epPCR applications than expected according to the in silico evaluation. Primer sets Mesa and Meth detected closely related organisms, which was a significant drawback as cross-reacting organisms were thermophilic and likely to inhabit similar habitats as members of the desired methanogenic genera do. Although the copy numbers added and calculated and the actually measured numbers were not congruent in our in vitro qPCR evaluation, the orders of magnitude reached proved that affiliated reference organisms were detected in much higher quantities than false-positive responding organisms. These results can be regarded as a quality criterion for accurate working primer sets in adapted qPCR applications. Based on the results we obtained by a comprehensive research approach, combining in silico, in vitro and in situ tests, we recommend the use of primer pairs Arc, MSL, Mecu, MBT and MCC in SYBR Green-based approaches for specific and accurate quantification of archaeal and methanogenic members of corresponding physiological groups, while the application of Mesa, Mesae, MMB and Meth primer sets is not suitable in SYBR Green assays.
Due to the highly specific and reliable performance of five out of nine primer sets, we applied them for in situ measurements in a typical methanogenic habitat—thermophilic fermenter sludge. Even though members of the Methanococcales have so far only been found in marine sediments (Liu and Whitman 2008), corresponding primers were evaluated in this study for potential application, although in an unlikely habitat, serving as out-group. The high congruence of qPCR and DGGE/sequencing results by investigating fermenter sludge samples convinced us of the specificity and accuracy of these sets in combination with our adaptions of the SYBR Green-based detection system. The occurrence of Methanomicrobiales (Methanoculleus sp.) and Methanosarcinales (Methanosarcina sp.) in significant abundances in the investigated habitat is additionally confirmed by previous studies (Lins et al. 2010; Malin and Illmer 2008; Wagner et al. 2011), supporting the qPCR-based quantification results. Even though we could not prove members of the Methanobacteriales at species level by DGGE/sequencing in the in situ measurements, while detecting them with qPCR, previous studies dealing with the same sludge were able to do so, detecting Methanothermobacter species in distinct abundances (Malin and Illmer 2008; Wagner et al. 2011), and supporting current MBT detection.
In this investigation we did not consider the fifth known class of methanogenic Archaea, the Methanopyrales, because their only approved habitat are marine geothermal sediments and growth temperatures of about 98 °C (Liu and Whitman 2008).
Due to the fact that all large investigated methanogenic groups (the Methanosarcinales, Methanomicrobiales and Methanobacteriales) include members that are well-known for (co-)inhabiting anaerobic digesters, cross-reactivity seems to be a central problem that might lead to significantly altered qualitative and quantitative results. Further information about environmental parameters, such as temperature, nutrient availability and phylogenetic affiliation, is a necessary prerequisite in order to decide whether the application of these primer pairs is appropriate.
Due to multiple operon copies in many organisms (Klappenbach et al. 2001) and limited information about methanogenic genomes (Yu et al. 2005), the determination of exact organismic numbers is also a challenge. Furthermore, operons can exist as identical copies or in different forms, sometimes with big sequence differences, which makes the interpreting the results even more difficult (Acinas et al. 2004). In some cases the change to functional gene quantification may be reasonable. However, such functional genes that are characteristic for a certain phylogenetic group are not always available or allow only a rough differentiation of a certain community. Even though in the case of methanogenic Archaea such genes are available (mcrA quantification) (Nunoura et al. 2008), we wanted a more detailed differentiation of this diverse community.
In conclusion, the practice of primer adaption from probe- to SYBR Green-based assays is valuable regarding cost reduction. However, a critical multifactorial evaluation of such adapted assays in silico, in vitro and in situ is essential. Furthermore, a more critical review and control process of commonly used 16S rRNA gene targeting primers regarding specificity and accuracy is needed. Primers are designed on basis of up to a current date status of online databases and may therefore become obsolete in future. However, we recommend to apply the in silico designed primers not only in combination with target organisms in vitro, but also with non-target organisms and to evaluate the appropriateness of a certain primer pair for every complex habitat they should be applied at. This is of course a challenging topic and we also came to limits we could so far not evaluate. Although, for current and future research on fermenter sludge, in combination with the designed post-analytical methodology, it is possible to draw conclusions about methanogenic developments in this certain habitat.
References
Abd-Elsalam K (2003) Bioinformatic tools and guideline for PCR primer design. Afr J Biotechnol 2:91–95
Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol 186:2629–2635
Anderson I, Ulrich L, Lupa B, Susanti D, Porat I, Hooper S, Lykidis A, Sieprawska-Lupa M, Dharmarajan L, Goltsman E, Lapidus A, Saunders E, Han C, Land M, Lucas S, Mukhopadhyay B, Whitman W, Woese C, Bristow J, Kyrpides N (2009) Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS ONE 4:1–9
Baker GC, Smith JJ, Cowan DA (2003) Review and re-analysis of domain-specific 16S primers. J Microbiol Meth 55:541–555
Blumer-Schuette S, Kataeva I, Westpheling J, Adams M, Kelly R (2008) Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr Opin Biotech 19:210–217
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Dam B, Kube M, Dam S, Reinhardt R, Liesacka W (2012) Complete sequence analysis of two methanotroph-specific repABC containing plasmids from Methylocystis sp. strain SC2. Appl Environ Microb 78:4373–4379
Diaz-Perez S, Alatriste-Mondragon F, Hernandez R, Birren B, Gunsalus R (1997) Bacterial artificial chromosome (BAC) library as a tool for physical mapping of the Archaeon Methanosarcina thermophila TM-1. Microb Comp Genomics 2:275–286
Einen J, Thorseth I, Ovreas L (2008) Enumeration of Archaea and Bacteria in seafloor basalt using real-time quantitative PCR and fluorescence microscopy. FEMS Microbiol Lett 282:182–187
Fogel G, Collins C, Li J, Brunk C (1999) Prokaryotic genome size and SSU rDNA copy number: estimation of microbial relative abundance from a mixed population. Microbial Ecol 38:93–113
Franke-Whittle IH, Goberna M, Insam H (2009) Design and testing of real-time PCR primers for the quantification of Methanoculleus, Methanosarcina, Methanothermobacter, and a group of uncultured methanogens. Can J Microbiol 55:611–616
Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li W, Liu J, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, de Macario EC, Ferry JG, Jarrell KF, Jing H, Macario AJL, Paulsen I, Pritchett M, Sowers KR, Swanson RV, Zinder SH, Lander E, Metcalf WW, Birren B (2002) The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res 12:532–542
Gregory T, Nicol J, Tamm H, Kullman B, Kullman K, Leitch I, Murray B, Kapraun D, Greilhuber J, Bennett M (2006) Eukaryotic genome size databases. Nucleic Acids Res 35:332–338
Illmer P, Wagner AO, Mair J, Malin C, Farbmacher S (2007) Chemical and biochemical parameters during composting of lawn clippings with special regard to the efficiency of a compost starter kit. Compost Sci Util 15:40–46
Klappenbach JA, Saxman PR, Cole JR, Schmidt TM (2001) rrndb: the ribosomal RNA operon copy number database. Nucleic Acids Res 29:181–184
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:1–11
Koonin E, Wolf YI (2008) Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res 36:6688–6719
Lins P, Malin C, Wagner AO, Illmer P (2010) Reduction of accumulated volatile fatty acids by an acetate-degrading enrichment culture. FEMS Microbiol Ecol 71:469–478
Lins P, Reitschuler C, Illmer P (2012) Development and evaluation of inocula combating high acetate concentrations during the start-up of an anaerobic digestion. Biores Technol 110:167–173
Liu Y, Whitman WB (2008) Metabolic, physiological, and ecological diversity of the methanogenic archaea. Proc Natl Acad Sci USA 1125:171–189
Luton PE, Wayne JM, Sharp RJ, Riley PW (2002) The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148:3521–3530
Malin C, Illmer P (2008) Ability of DNA-content and DGGE analysis to reflect the performance condition of an anaerobic biowaste fermenter. Microbiol Res 163:503–511
Maus I, Wibberg D, Stantcheff R, Eikmeyer F, Seffner A, Boelter J, Szczepanowski R, Blom J, Jaenicke S, König H, Pühler A, Schlütera A (2012) Complete genome sequence of the hydrogenotrophic, methanogenic archaeon Methanoculleus bourgensis strain MS2T, isolated from a sewage sludge digester. J Bacteriol 194:5487–5488
Murrell JC, Jetten MSM (2009) The microbial methane cycle. Environ Microbiol Rep 1:279–284
Muyzer G, DeWaal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nunoura T, Oida H, Miyazaki J, Miyashita A, Imachi H, Takai K (2008) Quantification of mcrA by fluorescent PCR in methanogenic and methanotrophic microbial communities. FEMS Microbiol Ecol 64:240–247
Pruesse E, Quast C, Knittel K, Fuchs B, Ludwig W, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:590–596
Sekiguchi H, Tomioka N, Nakahara T, Uchiyama H (2001) A single band does not always represent single bacterial strains in denaturing gradient gel electrophoresis analysis. Biotechnol Lett 23:1205–1208
Shigematsu T, Tang Y, Kawaguchi H, Ninomiya K, Kijima J, Kobayashi T, Morimura S, Kida K (2003) Effect of dilution rate on structure of a mesophilic acetate-degrading methanogenic community during continuous cultivation. J Biosci Bioeng 96:547–558
Skillman LC, Evans PN, Naylor GE, Morvan B, Jarvis GN, Joblin KN (2004) 16S ribosomal DNA-directed PCR primers for ruminal methanogens and identification of methanogens colonising young lambs. Anaerobe 10:277–285
Tatara M, Makiuchi T, Ueno Y, Goto M, Sode K (2008) Methanogenesis from acetate and propionate by thermophilic down-flow anaerobic packed-bed reactor. Biores Technol 99:4786–4795
Vallone P, Butler J (2004) AutoDimer: a screening tool for primer-dimer and hairpin structures. Biotechniques 37:226–231
Wagner AO, Malin C, Lins P, Illmer P (2011) Effects of various fatty acid amendments on a microbial digester community in batch culture. Waste Manage 31:431–437
Yu Y, Lee C, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89:670–679
Yu Y, Kim J, Hwang S (2006) Use of real-time PCR for group-specific quantification of aceticlastic methanogens in anaerobic processes: population dynamics and community structures. Biotechnol Bioeng 93:424–433
Yun JJ, LE Heisler LE, Hwang IIL, Wilkins O, Lau SK, Hyrcza M, Jayabalasingham B, Jin J, McLaurin J, Tsao MS, Der SD (2006) Genomic DNA functions as a universal external standard in quantitative real-time PCR. Nucleic Acids Res 34:1–10
Zhang T, Fang H (2006) Applications of real-time polymerase chain reaction for quantification of microorganisms in environmental samples. Appl Microbiol Biot 70:281–289
Zhu J, Zheng H, Ai G, Zhang G, Liu D, Liu X, Dong X (2012) The genome characteristics and predicted function of methyl-group oxidation pathway in the obligate aceticlastic methanogens, Methanosaeta spp. PLoS ONE 7:1–9
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Reitschuler, C., Lins, P. & Illmer, P. Primer evaluation and adaption for cost-efficient SYBR Green-based qPCR and its applicability for specific quantification of methanogens. World J Microbiol Biotechnol 30, 293–304 (2014). https://doi.org/10.1007/s11274-013-1450-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-013-1450-x