
Abstract
The intestinal microbiota has received increasing attention, as it influences growth, feed conversion, epithelial development, immunity as well as the intrusion of pathogenic microorganisms in the intestinal tract. In this study, pyrosequencing was used to explore the bacterial community of the intestine in gibel carp (Carassius auratus gibelio), and the origin of these microorganisms. The results disclosed great bacterial diversities in the carp intestines and cultured environments. The gibel carp harbored characteristic intestinal microbiota, where Proteobacteria were predominant, followed by Firmicutes. The analysis on the 10 most abundant bacterial operational taxonomic units (OTUs) revealed a majority of Firmicutes in the intestinal content (by decreasing order: Veilonella sp., Lachnospiraceae, Lactobacillales, Streptococcus sp., and Lactobacillus sp.). The second most abundant OTU was Rothia sp. (Actinobacteria). The most likely potential probiotics (Lactobacillus sp., and Bacillus sp.) and opportunists (Aeromonas sp., and Acinetobacter sp.) were not much abundant. Bacterial community comparisons showed that the intestinal community was closely related to that of the sediment, indicating the importance of sediment as source of gut bacteria in gibel carp. However, 37.95 % of the OTUs detected in feed were retrieved in the intestine, suggesting that food may influence markedly the microbiota of gibel carp, and therefore may be exploited for oral administration of probiotics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microorganisms formed spatially-organized communities as early as 3.25 billion years ago, and they colonized the digestive tract during the evolution of animals, becoming thus an integral part of the digestive system (Ley et al. 2008; Rawls et al. 2004). Studies on the intestinal microbiota of fish have revealed that the bacterial community may influence growth, feed conversion, epithelial development, and immunity of the host, as well as the settlement of pathogenic microorganisms in the intestinal tract (Nayak 2010; Ringø et al. 2010; Verschuere et al. 2000). Further characterization of intestinal microbiota is a prerequisite to understanding the structure of the bacterial community, and to identifying the features with potential benefits to fish health and rearing performance.
The intestinal microbiota of fish has been investigated by many researchers with conventional techniques, by using either culture-dependent, or culture-independent methods, like denaturing gradient gel electrophoresis (DGGE) and 16S rDNA clone libraries. These studies have demonstrated the specificity of intestinal microbiota in fish, with the predominance of Proteobacteria, but the bacterial composition may vary with age, individuals, developmental stage, nutritional status, environmental conditions, and the complexity of the fish digestive system (Kim et al. 2007; Nayak 2010; Wu et al. 2010). However, the proportion of bacteria that could be cultivated by conventional culture-dependent techniques was estimated between 0.1 and 10 % (Amann et al. 1995). There are also limitations in gene sequencing with conventional culture-independent methods, which impair the comprehension of the complexity of fish intestinal microbiota. Recently, the next-generation high-throughput DNA sequencing techniques were applied to study intestinal samples of zebrafish (Roeselers et al. 2011). These techniques threw new light on researches of intestinal microbiota in fish, and they revealed an unexpectedly high bacterial diversity in fish intestine.
The main purpose of surveying intestinal bacterial community is to find ways to manage its composition. In particular, it is crucial to determine the origin of fish gut bacteria. Cahill (1990) summarized that the bacteria present in fish gut resembled generally those observed in the environment or in the diet, and able to survive and to multiply in the intestinal tract. Ringø and Strom (1994) noticed that Enterobacteriaceae were dominant in the intestinal content of Artic charr, Salvilinus alpinus, fed with a capelin roe diet, while they recorded the predominance of Aeromonas sp. or Vibrio sp. in the fish fed with a commercial diet, and reared in freshwater or seawater, respectively. Ringø et al. (2006) reported that the viable counts of aerobic heterotrophic bacteria varied with respect to the diet in the gastrointestinal tract of healthy Atlantic cod, Gadus morhua. Ringø and Birkbeck (1999), and Romero and Navarrete (2006) concluded that the establishment of a balanced gut microbiota in fish larvae and fry resulted from the complex influence of bacteria from the egg, the live feed, and the rearing medium. However, most of these results were obtained under laboratory conditions—possibly biased to some extent—and the origin of fish gut microbiota in natural conditions remained uncertain.
Crucian carp, Carassius auratus, is one of the oldest cultured fish in the world, which is mainly produced in China. Limited investigations on the intestinal microbiota of goldfish (C. auratus) and gibel carp indicated that members from Proteobacteria and Firmicutes were dominant, and species of the genera Aeromonas, Acinetobacter, Bacillus, Cetobacterium, Clostridium, Lactococcus, Pseudomonas, Shewanella, Staphylococcus, Streptococcus, and Vibrio were common (Asfie et al. 2003; Feng et al. 2008; Silva et al. 2011). However, an in-depth study was still needed to comprehend the microbial diversity in the intestine of this important species for aquaculture. The present work was designed to characterize the intestinal microbiota of gibel carp by using pyrosequencing. In addition, water, sediment and fish feed samples were also analyzed, in an attempt to elucidate the origin of microorganisms in the intestine.
Materials and methods
Sample collection
The gibel carp were cultured in an artificial pond in Shishou City, Hubei Province, China from June 8 to November 15, 2010. During the experimental period, the fish were fed with commercial feed from Haida Co., Ltd., Jingzhou. At the end of the rearing experiment, two individuals with an average weight of approximate 500 g were caught. The fish were euthanazied by a high dose of anaesthetic (Tricaine methanesulfonate, Finquel MS 222, Sigma Inc). The skin of the fish was rinsed with 70 % ethanol to reduce contamination. The ventral surface was opened with sterile scissors to expose the body cavity, and thereafter, the intestine was sampled. The intestinal content of each fish was squeezed out and separately harvested. In addition, surface sediment samples were separately collected from 3 locations in the same pond, and pooled together. The water was sampled at a depth of approximate 50 cm from 3 locations in the pond, and pooled together. The microorganisms present in 250 mL water were concentrated onto a 0.2 μm filter. Fish feed was also taken for analysis. All the samples were provisionally stored in a portable refrigerator at −20 °C, transferred to laboratory within 24 h, and kept frozen at −80 °C until DNA extraction.
Isolation of bacterial DNA
Bacterial genomic DNA was extracted using a QIAamp® DNA Stool Mini Kit (Qiagen, Germany) with slight modification. The addition of sterile zirconia beads to the samples was tested on one intestinal content sample, which was extracted six times, either with (JYNa1, JYNa2 and JYNa3), or without bead beating (JYNb1, JYNb2 and JYNb3). For the other samples, DNA was extracted in duplicates, and the extracts from the same sample were pooled together to minimize the bias in sampling and extraction (Wu et al. 2010). The extracted DNAs were resuspended in TE buffer (pH 8.0) containing DNase-free RNase (TaKaRa Corporation Ltd., Dalian, China). Then, DNA purity and concentration were analyzed spectrophotometrically using the e-Spect ES-2 (Malcom, Japan), and the samples were stored at −20 °C.
PCR amplification, DGGE and T-RFLP (terminal-restriction fragment length polymorphism) analyses
To perform the DGGE analysis, a nested PCR was performed to target the V6–V8 region of the 16S rRNA gene. The primers 27F-1492R were used for the first PCR round, and 968F-1401R, with a GC clamp attached to the 5′-end of the 968F primer, served as the secondary primers (Heuer et al. 1997; Weisburg et al. 1991). The PCR reaction mix contained bacterial genomic DNA or PCR products (c. 5 ng), the primers (0.4 μM each), dNTPs (0.2 mM each), 1× PCR-buffer, Ex Taq DNA polymerase (0.5 U; TaKaRa Corporation Ltd., Dalian, China), and double-distilled water to a final volume of 25 μL. PCRs were performed using a TC-512 automated thermal cycler (Techne, UK). After 4 min of initial denaturation at 94 °C, the following conditions were applied: 30 s denaturation at 94 °C; 30 s annealing at 53 °C for the first round, and 57 °C for the second round; 90 s elongation for the first round, and 40 s for the second round, at 72 °C with 29 additional cycles, and a final 6 min extension at 72 °C. The PCR reactions were performed three times, and the products of each sample were mixed together for preventing bias in amplification.
Denaturing gradient gel electrophoresis profiling was performed on a Dcode universal mutation detection system (Bio-Rad laboratories Inc., USA) according to the manufacturer’s instructions. The PCR products were loaded onto 8 % (w/v) of polyacrylamide gels that contained 42–60 % denaturing gradient. The electrophoresis was run at 60 °C in 1× TAE buffer for 16 h at a constant voltage of 80 V. After electrophoresis, the polyacrylamide gels were stained for 30 min with silver nitrate, and photographed.
The 27F-1492R primers, with carboxifluorescein (5′-/6-FAM) labeled on the 5′-end of primer 27F, were also used for T-RFLP analysis. The amplification conditions were as listed above, but Blend Taq-plus polymerase (Toyobo, Japan) was used instead of Ex Taq polymerase. The PCR products of each sample were firstly digested with Mung bean nuclease (TaKaRa Corporation Ltd., China) to remove the single-stranded extensions. The digestion products were then purified using a DNA purification Kit (Axygen, China).
The T-RFLP analyses were performed in triplicates. Approximately 200 ng of the 16S rRNA gene amplification product from each sample was digested with restriction endonucleases HaeIII (15 U), MspI (10 U), or HhaI (10 U) (Fermentas, China) for 15 min at 37 °C. The digested fragments were precipitated using 70 % ethanol, and the efficiency of restriction digestion was detected by agarose gel electrophoresis. The fluorescent fragments were separated on a 3730 DNA Analyzer (Applied Biosystems, USA), and the sizes of each fragment were determined by comparison with the internal GeneScan™ 500 LIZ® Size Standard.
The terminal restriction fragments (TRFs) were visualized after electrophoretic separation using GeneMapper® Software v4.1 (Applied Biosystems, USA) and retrieved using the following criteria: the TRF sizes should lie between 50 and 500 bp, TRFs with fluorescent units (FU) not exceeding 200 were excluded, and two peaks were regarded as the same if the difference in peak sizes was less than 1 nt. A 0.5 % relative abundance threshold was also applied, and the TRFs with areas less than the threshold were removed from the remaining analyses. Only those TRFs present in two of the three independently-replicated electrophoreses of each digestion products were considered as valid TRFs.
PCR amplification, amplicon quantitation, pooling, and pyrosequencing
For each sample, the extracted DNA was used as template in the amplification of the V1–V3 region of the 16S rRNA gene. The bar-coded broadly conserved primers 27F and 533R containing the A and B sequencing adaptors (454 Life Sciences) were used to amplify this region. The forward primer (B-27F) was 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGAGAGTTTGATCCTGGCTCAG-3′, where the sequence of the B adaptor is shown in italics and underlined. The reverse primer (A-533R) was 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGNNNNNNNNNNTTACCGCGGCTGCTGGCAC-3′, where the sequence of the A adaptor is shown in italics and underlined and the Ns represent the unique eight base barcode used to distinguish PCR product from different samples. The 50 μL of PCR mix contained 0.6 μM each of the primer, c. 5 ng of template DNA, 0.2 mM of each dNTP, 1× PCR reaction buffer, and 2.5 U of Pfu DNA Polymerase (MBI. Fermentas, USA). The PCRs were run in a PTC-100TM Programmable Thermal Controller (MJ Research, Inc., Watertown, USA) using the following program: 4 min denaturing at 94 °C followed by 25 cycles of 30 s at 94 °C (denaturing), 30 s at 55 °C (annealing), and 30 s at 72 °C (elongation), with a final extension at 72 °C for 10 min. Three independent PCR reactions were performed for each sample. During reaction, negative controls were also performed. The triplicates of PCR products from the same sample were pooled together. The mixtures were visualized on agarose gels (2 % in TBE buffer) containing ethidium bromide, and purified with a DNA Gel Extraction Kit (Axygen, China).
Before sequencing, the DNA concentration of the PCR product of each sample was measured by using a Quant-iT PicoGreen double-stranded DNA assay (Invitrogen, Germany), and their quality was controlled on an Agilent 2100 bioanalyzer (Agilent, USA). After quantitation, the amplicons from each reaction mixture were pooled in equimolar ratios and subjected to emulsion PCR to generate DNA library, as recommended by 454 Life Sciences. Amplicon pyrosequencing was performed from the A-end using a 454/Roche A sequencing primer kit on a Roche Genome Sequencer GS FLX Titanium platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.
Bioinformatic and statistical analysis
For DGGE and T-RFLP data, the presence/absence of bands was exported to generate a matrix. Jaccard’s similarity coefficients were considered for the cluster analyses based on the DGGE and T-RFLP patterns, and determined with the program MVSP 3.1 (Kovach 1999).
Control standards were used to improve pyrosequencing data quality, and the valid reads were selected according to the following rules: 1, each read containing a primer sequence should be 350–600 bp in length; 2, with no ambiguous bases; 3, match the primer; 4, being one of the used barcode sequences; 5, at least 80 % of a previously determined 16S rRNA gene sequence. Otherwise, the read was regarded as artefact. The valid sequences were simplified using the ‘unique.seqs’ command to get a unique set of sequences, and then they were aligned using the ‘align.seqs’ command and compared with the Bacterial SILVA database (SILVA version 106; http://www.arb-silva.de/documentation/background/release-106/). The aligned sequences were further trimmed and the redundant reads were eliminated using the ‘screen.seqs’, ‘filter.seqs’, and ‘unique.seqs’ commands in order. Then, the ‘dist.seqs’ command was performed, and unique sequences were clustered into operational taxonomic units (OTUs), defined at the 97 % similarity threshold. Thereafter, the Shannon, Chao1, and ACE diversity indices were calculated. The rarefaction analysis and Good’s coverage for these libraries were determined. Heatmap figure, Venn diagram, and species rank abundance distribution curve (Whittaker plots) were generated using custom Perl scripts. In addition, a principal component analysis (PCA) was performed based on weighted UniFrac distance. In the present study, data preprocessing, OTU-based analysis, and hypothesis testing were performed on Mothur (Schloss et al. 2009).
Results
In total, 143,126 valid nucleotide sequences and 19,216 OTUs were retrieved from the ten samples through 454 pyrosequencing. Ninety reads and 64 OTUs were eukaryotic, and they were excluded from the following analyses. These sequences/OTUs were assigned to 25 different phyla/groups, and they were available through the NCBI/EBI/DDBJ Short Read Archive (accession number DRA000477; http://trace.ddbj.nig.ac.jp/DRASearch/submission?acc=DRA000477).
Effect of different methods of DNA extraction
The DNA extraction method with bead beating yielded 6,598 OTUs with an average 2,199 per replicate from the intestinal sample, while the method without bead beating produced 5,936 OTUs with a mean 1,978 per replicate. The independent t test was used to compare the two groups, and the result indicated that the difference was not significant (p = 0.1257).
Diversity analysis
In this study, 10,397–18,454 reads and 558–2,869 OTUs were retrieved from the ten samples (Fig. 1a). The highest species richness was observed in the sediment community, while the lowest one was detected in the PHSL community. The richness indices of ACE and Chao1 varied between 1,300 and 10,196, and between 1,025 and 6,467, respectively. The Shannon diversity index was between 3.06 and 6.53, with the lowest value in PHSL library and the highest one in CSLDN library (Fig. 1b). The rarefaction curve did not tend to reach the saturation plateau, except in the PHSL library (Fig. 2). Good’s estimation was used to determine the sequence coverage. It disclosed that 85–98 % of the species were observed in all the samples, with the lowest value in CSLDN community and the highest value in PHSL community (data not shown). The species rank abundance curves showed that only a few dominant OTUs were present in the different communities, and the majorities were rare ones (Fig. 3).

Alpha diversity of bacteria associated with gibel carp. a Number of OTUs observed and predicted (ACE and Chao1) and b Shannon diversity index. OTUs are grouped at 97 % similarity

Rarefaction curve of bacterial 16S rRNA sequences for different environmental samples. OTUs are identified using 97 % cutoffs

Rank abundance distribution curve showing the OTUs within each category of the Venn plot in Fig. S2 ranked according to their abundance in the corresponding combined OTU sequence data set
Taxonomic composition
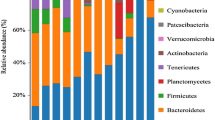
The bacterial patterns were quite dissimilar, even at the phylum level (Fig. 4). The CSLDN community consisted of the maximum number of phyla (23), and Firmicutes, Proteobacteria, and Bacteroidetes were the most important groups, amounting to 78.24 % of the total reads. The CSLW community was composed of 16 phyla, and Proteobacteria, Actinobacteria and Bacteroidetes were dominant, accounting for 84.09 % of the reads. The PHSL community had the simplest bacterial diversity, which was dominated by sequences related to Cyanobacteria (74.61 %), followed by Firmicutes (8.30 %), and Bacteroidetes (7.78 %). Some variability was observed between individuals and between replicates of the intestinal samples (JYNC, JYNa and JYNb). However, Proteobacteria and Firmicutes were predominant, representing 53.85–67.13 % of the reads of different JYN samples. In addition, the sequences related to Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Fusobacteria, Planctomycetes and Verrucomicrobia were above the abundance threshold (1 %) in all of the intestinal samples.

Relative abundance of different bacterial phyla within the different communities. Sequences that could not be classified into any known group were assigned as ‘Unclassified bacteria’
The most abundant OTUs (97 % similarity) varied among the different samples. The CSLDN library included sequences mostly similar to Veillonella (4.48–5.03 %), Prevotella (1.00–3.72 %), Lactobacillus (3.17 %), Bacteroides (1.43 %), and Dechloromonas (1.17–1.28 %) (Table 1). The CSLW library was dominated by sequences most closely related to Rhodocyclaceae (2.33–8.33 %), Neisseria (3.80 %), Microbacteriaceae (2.27–3.09 %), Cyanobacteria (2.21 %), and Limnohabitans (2.11 %) (Table 1). The most abundant OTUs in the PHSL library were those sequences similar to Cyanobacteria (1.86–42.14 %), Veillonella (1.80 %), Fusobacterium (1.13 %), Actinomyces (1.00 %), Anaeroglobus (0.84 %), Prevotella (0.79 %) and Lactobacillus (0.78 %) (Table 1). The ten most abundant OTUs associated with JYN libraries were different between individuals and even among replicates from the same individual with the same or different DNA extraction method. The JYNC library was dominated by sequences related to Veillonella (15.30 %), followed by Rothia (8.97 %) and Lachnospiraceae (4.73 %), while the intestinal libraries obtained for the second individual were dominated by sequences similar to Lactobacillales (unspecified; 2.42–6.95 %), Streptococcus (1.23–5.16 %), Lactobacillus (1.82–2.73 %) and Prevotella (1.48–1.63 %) (Table 1).
Species from the genera Lactobacillus, Lactococcus, Bacillus and Bifidobacterium are common sources of probiotics (Ringø et al. 2010; Verschuere et al. 2000). In this study, sequences related to genera Lactobacillus, Lactococcus and Bacillus were poorly abundant in all JYN communities, except that Lactobacillus in JYNa1 and JYNa2 libraries (10.03 and 4.57 %, respectively). The genus Bifidobacterium was only detected in JYNa1 library, where it corresponded to 0.01 % of the total reads (Table 2). In addition, nearly all the JYN samples included a low abundance of sequences related to Aeromonas and Acinetobacter (Table 2).
Similarity between the environmental microbiota and the intestinal content
The bacterial DGGE analysis showed band patterns that there were markedly different between JYN samples and the other samples. Further hierarchical cluster analysis using MVSP 3.1 disclosed that the JYN samples grouped together, while CSLW and CSLDN samples formed another clade (Fig. 5). The T-RFLP data confirmed this cluster pattern (data not shown).

Cluster analysis of 16S rRNA banding profile for bacterial communities from different environmental samples
Besides DNA-fingerprinting methods, the pyrosequencing data were used for creating a deep molecular inventory of microbiota in the different samples. The PCA score plot was determined by using the weighted UniFrac distance. The JYN libraries clustered to the right of the graph along the primary axis (66.06 % of the total variability) and remained distinct from the environmental libraries (Fig. 6). The CSLW and CSLDN libraries also grouped together, and they were distant from the PHSL library (Fig. 6). Overall, the two PCA axes explained 77.41 % of the variation between the different libraries. The hierarchically clustered heatmap analysis indicated that JYN libraries grouped firstly together, and then they clustered with CSLDN, PHSL and CSLW libraries, by decreasing order of similarity (Fig. S1).

The score plot of principal component analysis for different bacterial communities using weighted UniFrac distance. Principal components (PCs) 1 and 2 accounted for 66.06 and 11.35 % of the variance, respectively
The bacterial species shared between the JYN communities and the other communities were analyzed by a Venn diagram, in an attempt to identify the origin of the intestinal microbiota. The JYN communities shared 763, 291, and 211 bacterial species with the CSLDN, CSLW, and PHSL communities, respectively (Fig. S2). Among these common species, Veillonella sp. was the most abundant (OTU322, representing 4.09, 5.03, 1.40, and 1.80 % of the JYN, CSLDN, CSLW, and PHSL communities, respectively). The PHSL community had the highest proportion of OTUs shared with the JYN communities (37.95 %). In total, 1,102 OTUs were common to the fish intestinal content and to the culture-associated environments, including feed (17.4 % of total OTUs). Further rank abundance plot depicted the OTUs within each category of the Venn diagram in figure S2 ranked according to their abundance in the combined 143,036 sequence data set, and showed that the shared OTUs were highly abundant (Fig. 3).
Discussion
Prior to the microbial analyses, two methods of DNA extraction were compared. The results did not show any significant difference between the two methods, though the addition of the bead-beating step tended to increase the number of OTUs. This finding supported Yu and Morrison (2004) that bead beating improved the extraction yield of DNA from digestive microbiota. There was still some variation in the bacterial community composition among replicates from the same intestinal sample with either DNA-extraction method, indicating some experimental bias. However, the clustering- and sorting-based analyses—namely, DGGE, heatmap and PCA analyses—revealed that the replicated analyses of the intestinal content from one individual grouped together, showing thus the reproducibility of the methods. In order to avoid overestimating the rare phylotypes with pyrosequencing, we followed the recommendations of Kunin et al. (2010), who emphasized the importance of stringent quality-based trimming of 16S pyrotags and clustering thresholds no greater than 97 % identity.
The present study disclosed high bacterial diversity in the intestinal content and in the culture environment of the fish. The bacterial community present in the sediment was more diverse in terms of OTU counts, Good’s coverage estimate, species richness, and Shannon’s entropy index, while the rarefaction curve did not reach the saturation plateau. These findings are consistent with the study of Lozupone and Knight (2007), who demonstrated that microbes in sediment are more phylogenetically diverse than any other environment type. However, Han et al. (2010) reported that microbiota was more diverse in the gut content than in the samples collected from the rearing environment of grass carp, Ctenopharyngodon idellus. This incongruence may be due to the difference in methodology. Han et al. (2010) used clone libraries, where only 100 clones for each sample were sequenced to characterize microbiota, which may limit the comparison to the most abundant phylotypes. The present work indicated that the intestinal libraries amounted between 1,652 and 2,501 OTUs, which is much higher than any of the previous investigations on the microbial diversity in fish intestinal content—for example, 48 OTUs in grass carp (Han et al. 2010), and 44 phylotypes in yellow catfish (Pelteobagrus fulvidraco) (Wu et al. 2010).
The present intestinal bacterial composition analysis revealed that Proteobacteria was predominant, followed by Firmicutes. Roeselers et al. (2011) confirmed that Proteobacteria, especially γ-Proteobacteria, were dominant in the intestinal microbiota of fishes living in sea and in freshwater, while the abundance of Firmicutes was highly variable in zebrafish, depending on the origin. Asfie et al. (2003), Feng et al. (2008) and Silva et al. (2011) also identified Proteobacteria and Firmicutes as the dominant phyla in the intestine of C. auratus. Although the present study was generally consistent with these findings at the phylum level, the bacterial genera detected by Asfie et al. (2003), Feng et al. (2008) and Silva et al. (2011) accounted for small proportions of the total reads in the present study (less than 0.5 %), except for Streptococcus (3.9 %) and Clostridium (1.0 %). The high dominance of some genera in the previous studies was likely due to culture-dependent methods of characterization. In natural environments, most microorganisms cannot be easily isolated and cultivated on traditional agar substrates (Amann et al. 1995; Van Elsas and Van Overbeek 1993), which resulting in biased results. However, the proportion of many genera may be subjected to natural changes. For example, the proportion of Aeromonas sp., Pseudomonas sp., and Streptococcus sp. may vary with the season (Lee et al. 2002; MacMillan and Santucci 1990; Zu et al. 2000).
Probiotics are useful as both biological control agents against fish disease and activators of nutrient intake by the host, and they have been widely used in aquaculture (Ringø et al. 2010; Verschuere et al. 2000). The aptitude to successful colonization of the intestine is often considered as prerequisite for dietary probiotics, and the candidate strains should come preferably from fish intestinal microbiota. In this study, Lactobacillus sp., Lactococcus sp., and Bacillus sp. were present in the two individuals investigated. Some of these culturable strains may be of particular interest as potential probiotics. Although some strains of Bifidobacterium sp. are used as probiotics in other animals (Ringø et al. 2010), there was not significant detection of sequences related to the genus Bifidobacterium in the intestinal samples of gibel carp, suggesting that bacteria of this genus may not be suitable as candidate probiotics. In addition, the genera Aeromonas and Acinetobacter were also poorly abundant in gibel carp intestine. Sequences related to genera Aeromonas and Acinetobacter have been demonstrated to be associated with bacterial septicemia and other bacterial fish diseases (Li and Huang 2003; Lu et al. 2010; Shi et al. 1998). Although these disease-associated bacteria are not necessary pathogens, they may be opportunistic colonizers of decomposing intestinal content. The presence of these bacteria suggests that fish intestine may be a potential pathogenic reservoir, which may cause disease outbreaks in fish exposed to the stressors encountered in intensive farming (Ringø and Birkbeck 1999). Among the 10 most abundant bacterial OTUs in the intestine, Lactobacillus sp. may be a potential probiotic, while Streptococcus sp. and Prevotella sp. could play key roles in food degradation (Russell et al. 1981; Xu and Gordon 2003).
Fishes live in a complex ecosystem. Nayak (2010) reviewed the rearing and farming conditions that are critical factors that affect intestinal bacterial colonization and the subsequent establishment process. The PCA analysis and heatmap figure disclosed that bacterial community of the sediment was the closest to that of the intestinal communities, indicating that microbiota of the sediment is the most important factor in determining the bacterial community of gibel carp gut. The phylotypes identified by Han et al. (2010) from the intestines of grass carp and those detected in the feed looked closer than those observed in the habitat, but the comparison was based on a limited number of sequences in the 16S rRNA clone libraries. Ringø et al. (2006) and Reid et al. (2009) noted that dietary bacteria may remarkably influence microbiota in fish intestine. According to our results, although the intestinal microbiota pattern was more similar to that of the sediment, 37.95 % of the OTUs identified in feed were retrieved in the intestine. In agreement with Verschuere et al. (2000), the feed of gibel carp may be therefore supplemented with probiotics. However, the fish intestinal content and the culture-associated environments including feed have only 17.4 % bacterial OTUs in common, supporting the concept of a characteristic intestinal microbiota in fish, distinct from those present in the feed and in the environment (Han et al. 2010; Romero and Navarrete 2006), and the core gut microbiota in fish may be Proteobacteria and Firmicutes.
References
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Asfie M, Yoshijima T, Sugita H (2003) Characterization of the goldfish fecal microflora by the fluorescent in situ hybridization method. Fish Sci 69:21–26
Cahill MM (1990) Bacterial flora of fishes: a review. Microb Ecol 19:21–41
Feng X, Wu ZX, Zhu DM, Wang Y, Pang SF, Yu YM, Mei XH, Chen XX (2008) Study on digestive enzyme-producing bacteria from the digestive tract of Ctenopharyngodon idellus and Carassius auratus gibelio. Freshw Fish 38:51–57
Han SF, Liu YC, Zhou ZG, He SX, Cao YA, Shi PJ, Yao B, Ringø E (2010) Analysis of bacterial diversity in the intestine of grass carp (Ctenopharyngodon idellus) based on 16S rDNA gene sequences. Aquac Res 42:47–56
Heuer H, Krsek M, Baker P, Smalla K, Wellington EMH (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63:3233–3241
Kim DH, Brunt J, Austin B (2007) Microbial diversity of intestinal contents and mucus in rainbow trout (Oncorhynchus mykiss). J Appl Microbiol 102:1654–1664
Kovach W (1999) MVSP-a multivariate statistical package for Windows, ver. 3.1. Kovach Computing services, Pentraeth
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P (2010) Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123
Lee C, Cho JC, Lee SH, Lee DG, Kim SJ (2002) Distribution of Aeromonas spp. as identified by 16S rDNA restriction fragment length polymorphism analysis in a trout farm. J Appl Microbiol 93:976–985
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6:776–788
Li XB, Huang WF (2003) Study on the bacterial septicemia of Carassius auratus of penze (♀) × Cyprinus actidorsalis (♂) I-isolation and identification of the pathogen. Microbiology 30:56–60
Lozupone CA, Knight R (2007) Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104:11436–11440
Lu WH, Chen H, Zou Y, Yang YJ, Wang XD, Huang CG (2010) Identification and drug sensitive test of the pathogen in Acinetobacter disease from hybrid crucian carp (Carassius auratus gibelio ♀ × Cyprinus carpio ♂). Fish Sci 29:156–161
MacMillan JR, Santucci T (1990) Seasonal trendy in intestinal bacterial flora of farm raised channel catfish. J Aquat Anim Health 2:217–222
Nayak SK (2010) Role of gastrointestinal microbiota in fish. Aquac Res 41:1553–1573
Rawls JF, Samuel BS, Gordon JI (2004) Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci USA 101:4596–4601
Reid HI, Treasurer JW, Adam B, Birkbeck TH (2009) Analysis of bacterial populations in the gut of developing cod larvae and identification of Vibrio logei, Vibrio anguillarum and Vibrio splendidus as pathogens of cod larvae. Aquaculture 288:36–43
Ringø E, Birkbeck TH (1999) Intestinal microflora of fish larvae and fry. Aquac Res 30:73–93
Ringø E, Strom E (1994) Microflora of arctic charr, Salvilinus alpinus (L). Gastrointestinal microflora of free living fish and effect of diet and salinity on intestinalmicroflora. Aquac Fish Manag 25:623–629
Ringø E, Sperstad S, Myklebust R, Refstie S, Krogdahl A (2006) Characterisation of the microbiota associated with intestine of Atlantic cod (Gadus morhua L.)—the effect of fish meal, standard soybean meal and a bioprocessed soybean meal. Aquaculture 261:829–841
Ringø E, Olsen RE, Gifstad TO, Dalmo RA, Amlund H, Hemre GI, Bakke AM (2010) Prebiotics in aquaculture: a review. Aquac Nutr 16:117–136
Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF (2011) Evidence for a core gut microbiota in the zebrafish. ISME J. doi:10.1038/ismej.2011.1038
Romero J, Navarrete P (2006) 16S rDNA-based analysis of dominant bacterial populations associated with early life stages of coho salmon (Oncorhynchus kisutch). Microb Ecol 51:422–430
Russell J, Bottje W, Cotta M (1981) Degradation of protein by mixed cultures of rumen bacteria: identification of Streptococcus bovis as an actively proteolytic rumen bacterium. J Anim Sci 53:242
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537
Shi CY, Zhu XL, Lu QZ (1998) Studies on the hemostatic disorder in crucian carp induced by bacterial septicemia. Acta Hydrobiol Sin 22:367–370
Silva FC, Nicoli JR, Zambonino-Infante JL, Kaushik S, Gatesoupe FJ (2011) Influence of the diet on microbial diversity of faecal and gastrointestinal contents in gilthead sea bream (Sparus aurata) and intestinal contents in goldfish (Carassius auratus). FEMS Microbiol Ecol 78:285–296
Van Elsas JD, Van Overbeek LS (1993) Bacterial responses to soil stimuli. In: Kjelleberg S (ed) Starvation in bacteria. Plenum Press, New York, pp 55–80
Verschuere L, Rombaut G, Sorgeloos P, Verstraete W (2000) Probiotic bacteria as biological control agents in aquaculture. Microbiol Mol Biol Rev 64:655–671
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Wu SG, Gao TH, Zheng YZ, Wang WW, Cheng YY, Wang GT (2010) Microbial diversity of intestinal contents and mucus in yellow catfish (Pelteobagrus fulvidraco). Aquaculture 303:1–7
Xu J, Gordon JI (2003) Honor thy symbionts. Proc Natl Acad Sci USA 100:10452–10459
Yu Z, Morrison M (2004) Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36:808–812
Zu GZ, Yu WY, Li JN (2000) Epidemiological investigation and diagnosis of the bacterial septicemia of grass carp (Ctenopharyngodon idellus). Freshw Fish 30:35–37
Acknowledgments
The research was financially supported by grants from National Natural Science Foundation of China (No. 31272706), National Basic Research Program of China (No. 2009CB118705), the Natural Science Foundation of Hubei Province (No. 2009CDB331), and the earmarked fund for China Agriculture Research System (No. CARS-46-08).
Author information
Authors and Affiliations
Corresponding author
Additional information
Shangong Wu and Jingyun Tian contributed equally to this work.
JYN, PHSL, CSLW, and CSLDN represent intestinal content of gibel carp, feed, water, and sediment, respectively. JYNa and JYNb stand for the same fish intestinal content, while JYNC represents another fish gut content.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11274_2013_1322_MOESM1_ESM.tif
Fig. S1 Hierarchically clustered heatmap of the bacterial distribution of different communities. Row represents the relative percentage of each bacterial family, and column stands for different samples. Clusterings based upon the distance of the different libraries along the X-axis and the bacterial families along the Y-axis are indicated in the upper and left of the figure, respectively. The relative values for each bacterial family are depicted by color intensity with the legend indicated at the upper left of the figure. The Z-score denoted a measure of distance, in standard deviations, away from the mean (TIFF 914kb)
Rights and permissions
About this article
Cite this article
Wu, SG., Tian, JY., Gatesoupe, FJ. et al. Intestinal microbiota of gibel carp (Carassius auratus gibelio) and its origin as revealed by 454 pyrosequencing. World J Microbiol Biotechnol 29, 1585–1595 (2013). https://doi.org/10.1007/s11274-013-1322-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-013-1322-4