
Abstract
Valsa mali var. mali (Vmm), is the predominant species of apple valsa canker in China. Modern analysis of genes involved in virulence or pathogenicity usually implicate gene expression analysis most often performed using real-time quantitative polymerase chain reaction (RT-qPCR). However, for relative gene expression analysis pertinent reference genes have to be validated before using them as internal reference. This has not been reported for Vmm, so far. Therefore, eight commonly used housekeeping genes (ACT, CYP, EF1-α, G6PDH, GAPDH, L13, TUB, and UBQ) were cloned and evaluated for their expression stability by geNorm and NormFinder. Overall, all of the candidate reference genes were found to be suitable for gene expression analysis. After analysis of 10 samples from different strains and abiotic stress treatments, G6PDH appeared to be the most suitable reference gene, whereas GAPDH was the least suitable. Moreover, taking G6PDH combined with L13 or CYP as reference genes, improved the reliability of RT-qPCR significantly. The influence of the reference system on expression data was demonstrated by analyzing Vmmpg-1 encoding an endo-polygalacturonase gene. Pectinases are considered key pathogenicity factors for this fungus. In order to better understand the role of pectinases in pathogenicity of Vmm, RT-qPCR was used for expression analysis. Our results may provide a guideline for future studies on gene expression of V. mali var. mali by using RT-qPCR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Valsa mali var. mali (Vmm), is the predominant variety of the causal agent of apple valsa canker in China (Wang et al. 2011). Apple valsa canker is a destructive disease that causes serious economic losses in eastern Asia (Sakuma 1990). The disease cannot be controlled effectively by chemical treatments (Abe et al. 2007) since the pathogen penetrates extensively into the host phloem and xylem where it is not accessible to conventional fungicide treatments (Tamura and Saito 1982). Up to now, effective disease management strategies could not be established mainly because of the limited information about the pathogenesis of Vmm. Cell wall degrading enzymes (CWDEs) secreted by the fungus, such as pectinases (Ke et al. 2013), might play an important role during the infection process. For instance, the pectinases secreted by Leucostoma cincta and L. persoonii, the causal agents of peach canker, are thought to be pathogenic factors (Traquair 1987; Svircev et al. 1991). In order to clarify the role of pectinases in the infection of Vmm, it is necessary to establish an effective gene expression analysis method.
Real-time quantitative polymerase chain reaction (RT-qPCR) is widely used for analyzing gene expression, since it is efficient, sensitive and reliable to quantify transcript levels (Bustin 2002). Quantitative examination of gene expression in Vmm during infection may help to identify genes involved in virulence, or even pathogenesis. In order to minimize experimental errors produced by the quantity and the integrity of the RNA templates, and the efficiency of cDNA synthesis (Expósito-Rodríguez et al. 2008), a suitable normalization is crucial (Huggett et al. 2005). The most widely accepted normalization is to include one, or a small number of so-called housekeeping genes as internal references. A reference gene is a stably-expressed gene that is experimentally verified in a given species and tissue under specific experimental conditions. However, evidence showed that transcript levels of commonly used reference genes (e.g. GAPDH, ACTIN) may also vary considerably in response to changes in experimental conditions and/or tissue types (Ruan and Lai 2007; Thellin et al. 1999). Therefore, for every analysis a suitable reference gene(s) for the specific set of chosen experimental conditions needs to be selected. Validations of internal reference gene stability have been performed in some fungi, including Hypocrea jecorina (Steiger et al. 2010), Saccharomyces cerevisiae (Teste et al. 2009), Aspergillus niger (Bohle et al. 2007) and Candida albicans (Nailis et al. 2006). However, no systematic screen for stably-expressed reference genes for RT-qPCR in Vmm has been performed. For the present studies, we selected eight typically used housekeeping genes as candidate reference genes, including actin (ACT), cyclophilin (CYP), elongation factor 1-alpha (EF1-α), tubulin (TUB), glucose-6-phosphate-dehydrogenase (G6PDH), glyceraldehyde-3-phosphate-dehydrogenase (GADPH), ribosomal protein L13 (L13), and ubiquitin (UBQ), to validate and develop a RT-qPCR method in Vmm under different experimental conditions.
Materials and methods
Strains and abiotic stress treatments
The following strains of V. mali var. mali (Vmm) were included in our analyses: 03-8, SXYL20, SXWG36, NXLW92 and SDQX120 (Wang et al. 2011). All cultures were transferred on PDA plates and incubated at 25 °C for 4 days in the dark. For abiotic stress treatment only isolate 03-8 was used. For pH stress, a pure culture of 03-8 was incubated on PDA plate with the pH adjusted to 8.0 for 4 days in the dark. Temperature stress was applied by incubating the culture at 15 or 32 °C, respectively. For starvation treatment, the culture was incubated on PDA for 10 days. For fungicide treatment, the culture was incubated on PDA supplemented with 0.01 μg/mL difenoconazole for 4 days. Mycelium from each experiment was collected, quickly frozen in liquid nitrogen and stored at −80 °C prior to RNA extraction.
RNA extraction and cDNA synthesis
Harvested mycelia were ground to fine powder in liquid nitrogen using pre-cooled mortar and pestle. Total RNA from the samples was extracted using Trizol™ Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and then treated with RNase-free DNaseI at 37 °C for 30 min. Purification with Trizol™ Reagent was repeated to remove DNaseI. Only RNA samples exhibiting an A260/A280 ratio of 1.8–2.0 and an A260/A230 ratio >2.0 were used for subsequent analyses. The integrity of RNA samples was further evaluated by electrophoresis on ethidium bromide-stained 1.0 % agarose gels. First strand cDNA synthesis was performed using the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Shenzhen, PRC) and oligo-dT primers, following the manufacturer’s instructions in a final volume of 20 μL. All cDNA samples were stored at −20 °C and diluted 10-fold prior to use in RT-qPCR analysis. Genomic DNA contamination in cDNA was checked by RT-qPCR (Paolacci et al. 2009) using the following primer pair (5′-TACACTGCCACCCAGAAGACC-3′ and 5′-GCGTCGAAGATGGAGGAGTT-3′), which spans a 75 bp intron within the GAPDH gene. The 75 bp intron was identified based on the cDNA sequence of GAPDH obtained in this work (KC248181).
Cloning the candidate genes
At the onset of this study no sequence information for the potential candidate genes from Vmm was available in the publicly accessible databases. After aligning nucleotide sequences from numerous ascomycetes, degenerate primers were designed to perform 3′-RACE as gene specific forward primers (Table 1). For 3′-RACE, the adaptor primer 5′-GAGTCAGCAGGAATTCCATCGATTTTTTTTTTTTTTTTTT-3′ was used to perform reverse transcription. The 3′-universal reverse primer 5′-GAGTCAGCAGGAATTCCATCG-3′ was used for subsequent PCR amplification (Frohman et al. 1988). Primer sequences and the sizes of the amplified fragments of the eight genes analyzed are listed in Table 1. After PCR, the fragments were cloned, sequenced, and analyzed by tBLASTx (Altschul et al. 1990).
Primer design and RT-qPCR analysis
Real-time quantitative polymerase chain reaction primer pairs were designed to amplify 80–250 bp amplicons covering the sequenced region of the Vmm genes using Primer Premier V5.0 (Premier Biosoft International, Palo Alto, CA). Melting temperatures were adjusted to lie between 58 and 60 °C with primer length ranging from 18 to 24 bp. Specificity of the primers was tested by performing PCR with cDNA as template. PCR products were analyzed on a 2 % agarose gel. For each primer pair, PCR efficiency estimates were derived from a standard curve generated from a dilution series of pooled cDNA. Based on the crossing point cycle number (Cp values) for all dilution points in a series, a standard curve was generated using linear regression. PCR efficiency (E) is given by the equation: Efficiency % = (10(−1/slope) − 1) × 100 %.
Real-time quantitative polymerase chain reaction amplification was performed on an ABI 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA) using a SYBR Green based PCR assay. Every sample was analyzed in triplicate. The reaction was performed in a total reaction mix including cDNA (1 μL), PCR primers (10 μM, 0.4 μL), and SYBR Premix ExTaq (2×, 5 μL; Takara Biomedical Technology [Beijing] Co., Ltd.) in a total volume of 25 μL. PCR conditions were as follows: 95 °C for 1 min, 40 cycles of 95 °C for 10 s, 60 °C for 10 s, 72 °C for 40 s. Finally, dissociation curves were generated by increasing the temperature from 65 to 95 °C. Primer sequences and amplicon characteristics are given in Table 2.
Determination of candidate reference gene expression stability
Cp values of all samples were transformed to relative quantities, and then exported to geNorm version 3.5 (Vandesompele et al. 2002) and NormFinder (Andersen et al. 2004) for gene expression stability analysis. The geNorm statistical algorithm calculates gene stability (M value) with the average of pairwise variations, relying on the principle that the expression ratio of two ideal reference genes should be identical in all samples. Vandesompele et al. (2002) recommended using 1.5 as cut-off. Genes with lowest M value are most stable and geNorm was also used to estimate the normalization factor (NF n ) through the geometric mean of expression levels of n best reference genes. NormFinder, another algorithm for identifying the optimal normalization gene(s) among a set of candidates, ranked the candidate reference gene expression stability for all samples with no sub-group determination. According to the analysis, the lowest stability value would be top ranked.
Normalization of the target gene
Endo-polygalacturonase is a member of the pectinase family. The enzyme specifically degrades pectin substrate by cleaving α-1,4-glycosydic bonds between two non-methylated galacturonic acid residues (Hoondal et al. 2002). The corresponding gene, Vmmpg-1, was cloned from Vmm (data not shown) and used as target to demonstrate the efficiency of the validated candidate reference genes in RT-qPCR. The amplicon of Vmmpg-1 generated by the primer pair Vmmpg-1-forward: 5′-AAATGTGACGCCCGAAACTG-3′ and Vmmpg-1-reverse: 5′-TGCTTCCGATGGTGACTCTG-3′ was also verified by sequencing and melting curve analysis as described for the reference genes. For normalization experiments, Vmm strain 03-8 was cultured in 0.5 L Erlenmeyer flasks in stationary culture with 200 mL Murashige-Skoog (Murashige and Skoog 1962) medium. Mycelium was harvested at 5, 10 and 15 days after inoculation, respectively. RNA extraction and cDNA synthesis were performed as described above. Vmmpg-1 expression was normalized using three different strategies: (1) the most stably expressed reference gene, (2) the two most stably expressed reference genes, and (3) the least stably expressed gene (Lee et al. 2010).
Results
Cloning of the candidate reference genes and amplification specificity and efficiency
Based on homology cloning and 3′-RACE, partial sequences of the eight candidate reference cDNAs were obtained. The amplicon length for these eight cDNAs ranged from 518 bp for UBQ to 1,299 bp for G6PDH (Table 1). Contaminations with genomic DNA were excluded by trying to amplify an intron sequence of GAPDH. Absence of a signal in these analyses was taken as evidence that the cDNA templates were free from contamination with genomic DNA. Agarose gel electrophoresis (Fig. 1a) and melting curve analysis (Fig. 1b) revealed that all primer pairs amplified a single PCR product of the expected size. Furthermore, sequence analyses of cloned amplicons revealed that all fragments were identical or nearly identical to the sequences used for primer design (data not shown). The amplification efficiencies for the eight candidate reference genes ranged from 89.66 % (GAPDH) to 104.25 % (CYP), and regression coefficients varied from 0.9861 (L13) to 0.9991 (EF1-α), respectively (Table 2).

Specificity of RT-qPCR and amplicon size. a Agarose gel electrophoresis showing specific RT-qPCR products of the expected size for each gene. M represents the DNA marker. b Melting curves for all genes analyzed
Expression stability of candidate reference genes
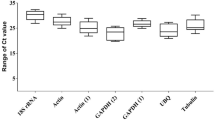
To reveal transcriptional differences among various candidate genes, the average Cp value was calculated across all samples. The Cp values for eight genes ranged from 21.73 to 24.27 (Fig. 2). Cp values were transformed to relative quantities using the delta-Cp method (Livak and Schmittgen 2001). geNorm (Vandesompele et al. 2002) and NormFinder (Andersen et al. 2004) were applied to calculate the expression stability of candidate reference genes.

RT-qPCR Cp values for reference genes. Expression data displayed as Cp values for each reference gene in all samples. Depicted is the median. Boxes indicate the 25 and 75 percentiles. Error-bars represent maximum and minimum values
Average expression stability (M value) of all genes was calculated by geNorm (Table 3). In our analyses, all genes showed an M value below the geNorm threshold of 1.5. Among the eight candidate genes, the ranking orders were similar in different sample series—G6PDH was the most stably expressed, while GAPDH was the least stably expressed. By ranking gene stability via stepwise exclusion of the least stable gene, geNorm identified the genes providing the lowest M value (Fig. 3). Among abiotic stress treatments (Fig. 3a), the best combination was G6PDH and L13. G6PDH and CYP were the most stable reference genes when different Vmm strains were analyzed (Fig. 3b). Taking both different strains and abiotic stress treatments into account (Fig. 3c), TUB and CYP were the most stable reference genes. To determine the optimal number of reference genes, geNorm was used to calculate the pairwise variation V n /V n+1 of two sequential normalization factors NF n and NF n+1. As reported by Vandesompele et al. (2002), a cut-off value of 0.15 was adopted. Among the different abiotic stress treatments, the combination of G6PDH and CYP produced a V2/3 value of 0.108 (Fig. 4a). In the strain comparison, two genes were sufficient for normalization, since the V2/3 value was <0.15 (Fig. 4b). Considering all samples, the V2/3 value was 0.165 and the V3/4 was 0.151, implicating that the involvement of the fourth gene was not necessary (Fig. 4c). Overall, G6PDH was demonstrated to be the ideal reference gene for all sets of samples.

Average expression stability values of reference genes by geNorm analysis. Gene expression values of eight genes across ten different treatments were obtained and analyzed using the geNorm software package. Genes were ranked by stepwise exclusion of the least stable gene. Genes with a high M value are less stably expressed compared with genes with a low M value. a different abiotic stress treatments, b different strains, c all samples

Determination of the optimal number of reference genes by geNorm analysis. Determination of the optimal number of reference genes for accurate normalization of gene expression. Average pairwise variations Vn/Vn+1 are calculated between the normalization factors NFn and NFn+1 to indicate whether inclusion of extra reference gene adds to the stability of the normalization factor. Every bar represents the change in normalization accuracy when stepwise adding more internal reference genes according to the ranking in Fig. 4. a different abiotic stress treatments, b different strains, c all samples
NormFinder can also analyze the stability of expression. The ranking generated by NormFinder was slightly different from that determined by geNorm (Table 3). Nevertheless, when applying NormFinder, G6PDH was still ranked the highest for all samples and different stresses, while L13 was the most stable for different strains. Similar to the results indicated by geNorm, NormFinder revealed that the least stable gene was GAPDH.
Evaluation of the expression of Vmmpg-1 by RT-qPCR
To demonstrate the efficiency of the validated reference genes in RT-qPCR, we analyzed the expression of Vmmpg-1 after different days of cultivation (Fig. 5). The relative expression level of Vmmpg-1 was normalized by one or two most stable reference gene(s), and the least stable gene. For strain 03-8, the relative expression level of Vmmpg-1 showed no significant differences when using either G6PDH alone or the combination of G6PDH and L13 as reference genes. However, GAPDH (the least stable reference gene) led to an underestimation of the Vmmpg-1 transcript level. These results further confirmed the significance of the validation of reference gene stability, which could effectively avoid the occurrence of low precision or unreliable results.

Relative quantification of Vmmpg-1 expression at different days after inoculation using validated reference genes for normalization. The validated reference gene(s), G6PDH, G6PDH + L13 and GAPDH, were used as normalization factors for analyzing Vmmpg-1 gene expression of Vmm strain 03-8. G6PDH and G6PDH + L13 were the most stable reference genes, while GAPDH was the least stable. Results are presented as a mean fold change in relative expression compared to the 15 dpi sampling stage
Discussion
In this study, we performed a series of experiments on the stability of eight commonly used housekeeping genes as internal controls for analyzing differential gene expressions in V. mali var. mali by RT-qPCR. As a fast, accurate and sensitive method for gene expression analysis, RT-qPCR has been widely applied in clinical diagnostics (Bustin and Dorudi 1998), gene expression studies in plants (Gachon et al. 2004) and fungi (Steiger et al. 2010; Teste et al. 2009; Bohle et al. 2007; Nailis et al. 2006). In order to get accurate data, one or more reference genes whose transcription levels are invariable under different experimental conditions are required as internal control. Normally, so-called housekeeping genes, such as ACT, or GAPDH are selected for normalization. However, recent evidence showed that the traditional housekeeping genes are not always stably expressed when tested under different experimental conditions and/or tissue types (Ruan and Lai 2007; Thellin et al. 1999). Obviously, the most suitable reference gene(s) should be verified for specific species with particular experimental conditions. We thus selected a series of candidate reference genes, including ACT, CYP, EF1-α, G6PDH, GAPDH, L13, TUB and UBQ. Since sequence information in databases online were lacking, partial cDNA sequences of each candidate reference gene were generate.
Besides the appropriate selection of reference genes, systematic errors (e.g. RNA extraction, cDNA synthesis, primer design) should be given attention when performing RT-qPCR analysis, since they might compromise the interpretation of results (Udvardi et al. 2008). To decrease systematic errors, a genomic DNA-finder primer pair spanning a 75 bp intron was designed to amplify GAPDH in all cDNA templates confirming the absence of genomic DNA. For gene-specific PCR primer design, the 3′-untranslated region is a good target, because it is generally more unique than coding sequences and closer to the start site of reverse transcription. Moreover, reverse transcription does not always extend to the 5′-end of the template. In our study, these modifications were taken into account.
In most cases, no single reference gene has a consistent expression level under different experimental conditions. Therefore, it is not suggested to find a universal reference gene. Basically, the application of two stable reference genes for normalization could produce more accurate and reliable data when compared to the application of a single reference gene. In the current study, taking the gene G6PDH combined with L13 or CYP as reference genes improved the reliability of RT-qPCR results.
Similar to the pertinent study, GPADH performed poorly among the most popular reference genes (Tong et al. 2009; Glare et al. 2002; Huggett et al. 2005). In the present study, GAPDH was demonstrated to be the worst gene in all sample pools by both geNorm and NormFinder analysis (Table 3). This means that traditional housekeeping genes may not be the best candidate reference genes for internal controls. However, due to the unavailability of gene sequences, only some highly conserved genes such as ACT, EF1-α can be easily sequenced, while the sequences of novel reference genes are variable among species which complicates the design of primers (Mariño-Ramírez et al. 2011). Mariño-Ramírez et al. (2011) demonstrated that transcriptome sequencing data is indeed a useful source of potential reference genes. Moreover, evidence showed that microRNAs had better expression stability than protein-coding genes (Feng et al. 2012). Therefore, commonly used housekeeping genes may not be the best choice if transcriptome sequencing data, microRNA sequencing data or other data are available.
Valsa mali var. mali, the causal pathogen of valsa canker on apple, causes serious economic losses in eastern Asia. Therefore, it is necessary to validate reference genes of RT-qPCR for this species. The RT-qPCR method described in this paper plays an important role in quantifying gene expression levels in Vmm, such as Vmmpg-1 expression. Therefore, we speculate that the evaluation of expression stability of Vmm reference genes might be useful in further molecular research on this species.
References
Abe K, Kotoda N, Kato H, Soejima J (2007) Resistance sources to valsa canker (Valsa ceratosperma) in a germplasm collection of diverse Malus species. Plant Breeding 126(4):449–453
Altschul Gish SF, Miller W, Myers W, Lipman EW, David J (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64(15):5245
Bohle K, Jungebloud A, Göcke Y, Dalpiaz A, Cordes C, Horn H, Hempel DC (2007) Selection of reference genes for normalisation of specific gene quantification data of Aspergillus niger. J Biotechnol 132(4):353–358
Bustin S (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29(1):23–39
Bustin S, Dorudi S (1998) Molecular assessment of tumour stage and disease recurrence using PCR-based assays. Mol Med Today 4(9):389–396
Expósito-Rodríguez M, Borges A, Borges-Pérez A, Pérez J (2008) Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol 8(1):131
Feng H, Huang X, Zhang Q, Wei G, Wang X, Kang Z (2012) Selection of suitable inner reference genes for relative quantification expression of microRNA in wheat. Plant Physiol Biochem 51:116–122
Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA 85(23):8998–9002
Gachon C, Mingam A, Charrier B (2004) Real-time PCR: what relevance to plant studies? J Exp Bot 55(402):1445–1454
Glare E, Divjak M, Bailey M, Walters E (2002) β-Actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels. Thorax 57(9):765–770
Hoondal G, Tiwari R, Tewari R, Dahiya N, Beg Q (2002) Microbial alkaline pectinases and their industrial applications: a review. Appl Microbiol Biotechnol 59(4):409–418
Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6(4):279–284
Ke X, Huang L, Han Q, Gao X, Kang Z (2013) Histological and cytological investigations of the infection and colonization of apple bark by Valsa mali var. mali. Australas Plant Pathol 42(1):85–93
Lee JM, Roche JR, Donaghy DJ, Thrush A, Sathish P (2010) Validation of reference genes for quantitative RT-PCR studies of gene expression in perennial ryegrass (Lolium perenne L.). BMC Mol Biol 11(1):8
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408
Mariño-Ramírez L, Demidenko NV, Logacheva MD, Penin AA (2011) Selection and validation of reference genes for quantitative real-time PCR in buckwheat (Fagopyrum esculentum) based on transcriptome sequence data. PLoS ONE 6(5):e19434
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15(3):473–497
Nailis H, Coenye T, Van Nieuwerburgh F, Deforce D, Nelis HJ (2006) Development and evaluation of different normalization strategies for gene expression studies in Candida albicans biofilms by real-time PCR. BMC Mol Biol 7(1):25
Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M (2009) Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol 10(1):11
Ruan W, Lai M (2007) Actin, a reliable marker of internal control? Clin Chim Acta 385(1–2):1–5
Sakuma T (1990) Valsa canker. In: Jones AL, Aldwinckle HS (eds) Compendium of apple and pear disease. American Phytopathological Society Press, St Paul, MN, USA, pp 39–40
Steiger MG, Mach RL, Mach-Aigner AR (2010) An accurate normalization strategy for RT-qPCR in Hypocrea jecorina (Trichoderma reesei). J Biotechnol 145(1):30–37
Svircev A, Biggs A, Miles N (1991) Isolation and partial purification of phytotoxins from liquid cultures of Leucostoma cincta and Leucostoma persoonii. Can J Bot 69(9):1998–2003
Tamura O, Saito I (1982) Histopathological changes of apple bark infected by Valsa ceratosperma (Tode ex Fr.) maire during dormant and growing periods. Ann Phytopath Soc Japan 48(4):490–498
Teste M-A, Duquenne M, François JM, Parrou J-L (2009) Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol Biol 10(1):99
Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E (1999) Housekeeping genes as internal standards: use and limits. J Biotechnol 75(2):291–295
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol 10(1):71
Traquair J (1987) Oxalic acid and calcium oxalate produced by Leucostoma cincta and L. persoonii in culture and in peach bark tissues. Can J Bot 65(9):1952–1956
Udvardi MK, Czechowski T, Scheible WR (2008) Eleven golden rules of quantitative RT-PCR. Plant Cell 20(7):1736–1737
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3(7):research0034
Wang X, Wei J, Huang L, Kang Z (2011) Re-evaluation of pathogens causing valsa canker on apple in China. Mycologia 103(2):317–324
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (NO.31171796 and NO.31101476), the Program for Agriculture (nyhyzx201203034-03) and the 111 Project (B07049). Special thanks are due to Dr. H. Buchenauer and Dr. Xiaoyu Qiang for correcting the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhiyuan Yin and Xiwang Ke contributed equally to this work.
Rights and permissions
About this article
Cite this article
Yin, Z., Ke, X., Huang, D. et al. Validation of reference genes for gene expression analysis in Valsa mali var. mali using real-time quantitative PCR. World J Microbiol Biotechnol 29, 1563–1571 (2013). https://doi.org/10.1007/s11274-013-1320-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-013-1320-6