
Abstract
A bacterial strain K9 capable of degrading malachite green was isolated from the sludge of the wastewater treatment system of a chemical plant. It was identified preliminarily as Pseudomonas sp. Strain K9 was also able to degrade other triphenylmethane dyes, such as Crystal Violet and Basic Fuchsin. The gene tmr2, encoding the triphenylmethane reductase, was cloned from strain K9, and functionally expressed in E. coli. A 5946-bp DNA fragment including the tmr2 gene was cloned from the genomic DNA of strain K9 by chromosome walking. Its sequence analysis showed that tmr2 was associated with a typical mobile element ISPpu12 consisting of tnpA (encoding a transposase), lspA (encoding a lipoprotein signal peptidase) and orf1 (encoding a putative MerR family regulator), orf2 (encoding a CDF family heavy metal/H+ antiporter). This association was also found in another malachite green-degrading strain Pseudomonas sp. MDB-1, which indicated that the tmr2 gene might be a horizontally transferable gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Malachite green (4-[(4-dimethylaminophenyl)-phenyl-methyl]-N, N-dimethylaniline, MG) is a triphenylmethane dye which has been widely used in textile industries for dyeing nylon, wool, silk, leather, and cotton (Parshetti et al. 2006; Daneshvar et al. 2007). MG has been also widely used as the most efficacious antifungal agent in the fish farming industry for its high activity against protozoal and fungal infections (Hoffman and Meyer 1974; Alderman 1985; Schnick 1988). Therefore, potential human exposure to MG could result from the consumption of treated fish and working in the dye and aquaculture industries. MG is highly toxic to mammalian cells; it promotes hepatic tumor formation in rodents and also causes reproductive abnormalities in rabbit and fish (Fernandes et al. 1991; Rao 1995). The structural similarity of MG to other carcinogenic triphenylmethane dyes such as Crystal Violet also raises suspicion of its carcinogenicity (Littlefield et al. 1985). The U.S. Food and Drug Administration nominated MG as a priority chemical for carcinogenicity due to its potential adverse effects to human health (Culp and Beland 1996), and the use of MG in the fish farming industry has been banned in several regions such as the EU, USA and other countries. However, it is still being used illegally as a practically indispensable disinfectant for its low cost, ready availability and antifungal effectiveness (Schnick 1988; European Commission 2006; Sudova et al. 2007). The physical and chemical properties of MG make it hard to be removed from water, and MG residue will eventually produce a potential threat to ecological balance and human health.
Several physico-chemical methods have been used to eliminate MG from water; but with intensive cost. Biological degradation is of great value as an inexpensive and less sludge-producing alternative to chemical decomposition (Utkarsha et al. 2008). Several microorganisms, including bacteria, yeast and fungi have been reported to biodecolorize triphenylmethane dyes (Wamik et al. 1998; Sani and Banerjee 1999; Cha et al. 2001; An et al. 2002; Liu et al. 2004; Eichlerova et al. 2006; Jadhav and Govindwar 2006; Ren et al. 2006; Parshetti et al. 2006; Utkarsha et al. 2008; Li et al. 2009). Laccase, peroxidase from fungi and triphenylmethane reductase (TMR) from bacteria are involved in the decolorization process. (Shin and Kim 1998; Tekere et al. 2001; Jang et al. 2005; Li et al. 2009).
In this study, a bacterial strain K9 capable of degrading MG and other triphenylmethane dyes has been isolated and characterized. Its triphenylmethane reductase-encoding gene tmr2 was cloned, sequenced and effectively expressed in E.coli BL21. The DNA fragment flanked the tmr2 gene was also cloned by chromosome walking, which was sequenced and analysed further.
Materials and methods
Media and chemicals
Mineral salts medium (MM) contained (g l−1) : NaCl, 1.0; NH4NO3, 1.0; KH2PO4, 0.5; K2HPO4, 1.5; MgSO4•7H2O, 0.1; pH7.0. MGM was MM supplemented with 20 mg MG l−1 as carbon source. LB medium contained (g l−1): tryptone; 10.0, yeast extract; 5.0; NaCl, 10.0; pH7.0.
Malachite green (purity 90%) was purchased from the Sigma–Aldrich Trading Co. Ltd., Shanghai, PR China. Crystal Violet, Basic Fuchsin, Neutral Red, Alizarin Red S, Orange II and other chemicals were of analytical grade and purchased from Shanghai Sangon Co. Ltd, PR China.
LA-Taq DNA polymerase and DNA restriction enzymes were purchased from Takara Biotechnology Co. Ltd., Dalian, PR China. Isopropyl-β-D-thiogalactopyranoside (IPTG) was purchased from Shanghai Genview Co. Ltd, PR China. Oligonucleotide synthesis and DNA sequencing were carried out by Invitrogen Biotechnology Co. Ltd., Shanghai, PR China.
Analytical method
For the determination of MG, 3 ml aliquots of the culture were sampled at different time intervals, centrifuged at 6,000 g for 10 min to eliminate the bacterial cells, MG contents of the supernatant was determined by an UV-visible scanning spectrophotometer (PC-2401) at the absorbance of 622 nm. The degradation rate was calculated from the difference between the initial and final absorbance values and expressed as a percentage of the original concentration. The detecting method for other dyes was similar to that of MG, the wavelengths for Crystal Violet, Basic Fuchsin, Neutral Red, Alizarin Red S and Orange II were 584, 542, 523, 508 and 484 nm, respectively.
Isolation and identification of MG-degrading strain
About 5 ml of activated sludge sample collected from the wastewater treatment system of a chemical plant was added into a 250-ml Erlenmeyer flask with 100 ml MGM. The culture was incubated at 30°C at 180 rev min−1 for 5 days. Five milliliters of enrichment culture was then subcultured into fresh MGM every five days. The degradation of MG in the enrichment cultures was determined, and appropriate dilutions of the enrichment culture with confirmed degrading effect were spread on MGM plates containing 2% (w/v) agar. After 4 days of incubation at 30°C, microbial colonies became visible and a transparent halo appeared around colonies capable of degrading MG, which were purified and determined for their degrading capability in liquid MGM.
The identification of strain K9 was carried out according to Bergey’s Manual of Determinative Bacteriology (Holt et al. 1994) and the sequence analysis of its 16S rRNA gene. The genomic DNA was extracted by the method of high-salt-concentration precipitation (Miller et al. 1988). The 16S rRNA gene was amplified by PCR using standard procedures. The PCR product was ligated into the vector pMD18-T and then transformed into E.coli DH5α for sequencing. The 16S rRNA gene sequence with 1,399 bp was deposited at GenBank under the Accession number EU855781. Alignment of the different 16S rRNA gene sequences from the GenBank database was performed using CLUSTALX 1.8.3 with default settings (Thompson et al. 1997). Phylogeny was analysed by MEGA version 3.0 software. Distances were calculated using the Kimura two-parameter distance model. The unrooted tree was built by the neighbor-joining method. The dataset was bootstrapped 1,000 times (Weisburg et al. 1991).
Degradation experiments
Strain K9 was pre-cultured in LB medium in a rotary shaker at 30°C and 180 rev min−1 overnight, and the cells were collected by centrifugation at 5,000 g for 5 min at room temperature. The cell pellets were washed twice with sterilized MM, and adjusted to an OD600 of 1.0. For degradation experiments, the cells were inoculated at 3% (v/v) level into 250-ml flask containing 100 ml MGM and then incubated at 30°C at 180 rev min−1. The culture samples were collected periodically for the determination of MG residual and the growth of strain K9.
Cross-feeding studies with other dyes by strain K9 were also performed. The liquid MM was supplemented with Crystal Violet, Neutral Red, Basic Fuchsin, Orange II and Alizarin red S at the concentration of 20 mg l−1, respectively.
Cloning and expression of the tmr2 gene from strain K9
The triphenylmethane reductase gene tmr2 was cloned from genomic DNA of strain K9 using a PCR-based technique. The primers TF and TR were designed according to the tmr2 gene (Li et al. 2009), which was shown in Table 1.The NdeI and XhoI sites (underlined) were incorporated into primers to facilitate directional cloning of the PCR product into (NdeI-XhoI sites of the expression vector) pET29a. Genomic DNA of strain K9 was extracted by the method described above and used as template. The PCR products were purified and ligated with pMD18-T vector. The resulting plasmid was designated as pMD18-T-tmr2, which was transformed into E. coli DH5α cells for the sequencing of the tmr2 gene. To express the tmr2 gene, the NdeI/XhoI fragment of pMD18-T-tmr2 was ligated with pET29a to form pET29a-tmr2, which was transformed into E. coli BL21(DE3). The transformants were subcultured into LB medium containing kanamycin (50 mg l−1) and grown until the culture turbidity reached 0.5 (OD600), then the tmr2 gene expression was induced by adding IPTG to the final concentration of 1 mM, then the 0, 1, 2, 3 h later protein extracts prepared from these cultures were analysed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Cloning of DNA fragment flanking the tmr2 gene
A simple chromosome-walking method SEFA-PCR (self-formed adaptor PCR)(Wang et al. 2007) was employed to clone the fragment upstream and downstream of tmr2 gene in strain K9 and Pseudomonas sp.MDB-1, respectively. Pseudomonas sp.MDB-1 was another MG-degrading strain harboring the tmr2 gene (Li et al. 2009). The primers used in this study are presented in Table 1. dSp1, dSp2 and dSp3 were used to amplify the downstream sequences of tmr2 gene in strain K9 and strain MDB-1, while uSp1, uSp2 and uSp3 were used to amplify the upstream sequences.
The general procedure of SEFA-PCR was as following: First, a single cycle PCR was carried out with Sp3 as primer, genomic DNA of strain K9 and MDB-1 as template, cycling conditions: (a) denaturing at 95°C for 90 s; (b) annealing at 30°C, then ramping to 70°C at 0.2°C/s (c) extension at 70°C for 5 min. Second, Sp1 was added to the reaction mixture and 25 cycles of PCR was carried out, cycling conditions: (a) denaturing at 94°C for 30 s; (b) annealing at 60°C for 30 s and extension at 70°C for 5 min. Third, 8 cycles of thermal asymmetric PCR was carried out, cycling conditions: (a) two cycles of denaturing at 94°C for 30 s, annealing at 60°C for 30 s and extension at 70°C for 5 min; (b) one cycle of denaturing at 94°C for 30 s, annealing at 50°C for 30 s, extension at 70°C for 5 min. Fourth, normal PCR was carried out with Sp2 as primer and 1μL of the above PCR products was used as template. Cycling conditions: (a) pre-denaturing at 95°C for 2 min; (b) 30 cycles of denaturing at 94°C for 30 s; annealing at 65°C for 30 s and extension at 72°C for 5 min; (c) extension at 72°C for 10 min. The PCR products were cloned to pMD18-T and subjected to DNA sequencing.
Results and discussion
Isolation and characterization of strain K9
After several times of enrichment, a bacterial strain K9 was isolated. It produced a transparent halo on the MGM solid plate. Taxonomic properties of strain K9 were as follows: a rod-shaped (1.3–1.6 × 0.6–0.8 μm); gram-negative; positive for oxidase activity, nitrate reduction and hydrolysis of starch; negative for indole test, Vogese-Proskauer test, Methyl-Red test, gelatin hydrolysis, phenylalanine deaminase test, urease test and citrate permease test. The 16S rRNA gene sequence of strain K9 showed high similarity with that of the Pseudomonas species. A phylogenetic tree based on known representatives of describing Pseudomonas species and other species is presented in Fig. 1. Based on the above characters, strain K9 was preliminarily identified as Pseudomonas sp.

Phylogenetic tree of strain K9 based on 16S rRNA gene sequence analysis. Bootstrap values obtained with 1,000 repetitions were indicated as percentages at all branches
MG degradation ability of strain K9
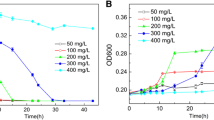
The kinetics of degradation of MG and growth of strain K9 were investigated simultaneously (Fig. 2). The logarithmic growth of strain K9 began at 6 h after inoculation. During the degradation, the growth showed a positive correlation with MG degradation. The cell number increased from 6 × 105 cells ml−1 to maximum 1.67 × 106 cells ml−1 at hour 42, and 88.30% of MG (20 mg l−1) was degraded within 48 h. The optimal growth temperature and initial pH for degradation of MG was 30°C and 7.0 respectively (data not shown). Many MG-degrading bacteria have been reported, Pseudomonas sp. MDB-1 could degrade more than 90% of MG (10 mg l−1) within 48 h (Li et al. 2009). Sphingomonas paucimobilis was able to degrade 75% of MG (50 mg l−1) within 4 h under shaking condition (Ayed et al. 2009). Citrobacter sp. could degrade 91% of 30 μM of MG within 1 h, which was the highest degrading efficiency ever reported. (An et al. 2002).

Kinetics of degradation of MG and growth of strain K9 a line with square MG concentrations of inoculated cultures; a line with triangle CFU of strain K9; a line with diamond MG concentrations in uninoculated control; the data are the means ± standard deviation for the triplicate treatments
The degradation of strain K9 on other dyes was also tested. It showed almost the same degrading activity against Basic Fuchsin and Crystal Violet as that of MG, indicating that strain K9 has high degradation efficiency on triphenylmethane dyes. But degradation capabilities of strain K9 on Neutral Red (quinoneimine), Alizarin Red S (anthraquinon) and Orange II (disazo) were very low, which were 11.81 ± 0.31%; 5.38 ± 0.12% and 10.03 ± 0.25%, respectively (Table 2).
Cloning and expression of the tmr2 gene of strain K9
The tmr2 gene was successfully cloned by PCR from strain K9, it was 864 bp in length (FJ649187), with high nucleic acid sequence similarity with triphenylmethane reductase gene (tmr) of Citrobacter sp. MY-5 (AY756172), triphenylmethane oxygenase gene (tpmD) of Aeromonas hydrophila subsp. strain DN322 (EF010984) and triphenylmethane reductase gene (tmr2) of Pseudomonas sp. MDB-1 (EF463103). The sequence similarity was 99, 99 and 100%, respectively.
The expression of tmr2 in E. coli BL21(DE3) strain was checked by SDS–PAGE. The result showed that tmr2 was overexpressed in E. coli BL21 with the induction of IPTG. The molecular weight of target protein was about 31KD (Fig. 3), which was similar to the expressed product of the tmr gene of Citrobacter sp. MY-5 (Jang et al. 2005) and the tmr2 gene in MDB-1 strain (Li et al. 2009). To determine the activity of the expressed product of the tmr2 gene, the induced E. coli BL21(DE3)/pET29a-tmr2 cells were used for the enzymatic catalysis of MG and Crystal Violet. The results showed that they could decolorize MG and Crystal Violet (Fig. 4), producing the same pattern as the tmr gene encoding enzyme of Citrobacter sp. MY-5 (Jang et al. 2005), which indicated the successful expression of tmr2 gene.

Expression of the tmr2 gene in E. coli. Lane M, molecular mass markers (in kDa); lane 1, protein extracts prepared from E. coli BL21 cells; lanes 2–5, protein extracts prepared from the cultures of E. coli BL21 cells with pET29a-tmr2 induced for 0, 1, 2and 3 h, respectively. Expression of tmr2 is indicated by an arrow

a Time-course of MG degradation by E. coli BL21/pET29a-tmr2b Time-course of Crystal Violet degradation by E. coli BL21/pET29a-tmr2
Cloning and analysing of sequences flanking the tmr2 gene
The tmr2 gene existed both in strain K9 and strain MDB-1 and had the same nucleic acid sequence. Therefore, the flanking sequences were analysed to determine whether this gene was horizontally transferable. The upstream and downstream fragments of the tmr2 gene of strain K9 and strain MDB-1 were cloned by SEFA-PCR with the primers listed in Table 1. Agrose gel electrophoresis of amplified products is shown in Fig 5. The sequence analysis revealed that, the length for upstream and downstream fragments of tmr2 gene of strain K9 was 1,076 bp and 4,006 bp respectively, while it was 1,411 bp and 3,698 bp for strain MDB-1. These fragments were spliced with the tmr2 gene fragment. The total length for the generated fragment of strain K9 (designated as F1) and strain MDB-1 (designated as F2) was 5,946 bp (GenBank Accession No. FJ649187) and 5,973 bp (GenBank Accession No. HM062496), respectively. Sequence analysis for F1 fragment of strain K9 is shown in Fig. 6. The tmr2 gene (1077-1940) of strain K9 was associated with a typical mobile element ISPpu12 (2155-5526) consisting of ISPpu12-IR (inverted repeat sequence; 2155–2178), orf1 (encoding a putative MerR family regulator; complement 2242–2649), orf2 (encoding a CDF family heavy metal/H+ antiporter; 2745–3641), lspA (encoding a lipoprotein signal peptidase; 3645–4157), tnpA (encoding a transposase; 4254–5468), ISPpu12-IR (inverted repeat sequence; 5503–5526). The association of the tmr2 gene with a typical mobile element ISPpu12 also existed in the F2 fragment of strain MDB-1. However, the left flanking sequence (designated as LF) of the association of strain K9 showed only 20% similarity with that of strain MDB-1. The right flanking sequence (designated as RF) of the association of strain k9 showed only 12% similarity with that of strain MDB-1. The mobile element ISPpu12 was also reported in several other bacteria generally associated with plasmids and xenobiotic degradation genes, and it had been suggested to be involved in gene silencing, activation, and horizontal transferring (Weightman et al. 2002). In the present study, the association of the tmr2 gene with ISPpu12 existed in two MG-degrading strains, which indicated that the tmr2 gene might be a horizontally transferable gene.

Agarose gel electrophoresis of SEFA-PCR products. M1: DL2000 marker; 1 upstream product of the tmr2 gene of strain MDB-1; 2 upstream product of the tmr2 gene of strain K9; 3 downstream product of the tmr2 gene of strain MDB-1; 4 downstream product of the tmr2 gene of strain K9; M2, λ-HindIII marker

Comparison of F1 fragment of strain K9 and F2 fragment of strain MDB-1 coding regions and transcriptional directions of the genes are indicated by broad arrows
Strain K9 showed high degradation efficiency against triphenylmethane dyes, but the degradation ability against dyes of other kinds, such as Neutral Red (quinoneimine), Alizarin Red S (anthraquinon) and Orange II (disazo) was very low (Table 2). Since tmr2 was a horizontally transferable gene, it could be introduced into strains with degrading ability against dyes other than triphenylmethane dyes in order to construct genetically engineered microorganisms. This could enhance the usage of tmr2 gene in wastewater treatment.
Conclusions
One MG-degrading strain Pseudomonas sp. K9 was isolated. It could also degrade other triphenylmethane dyes, indicating its potential usage in the treatment of wastewater from dye industries. The gene tmr2, encoding the triphenylmethane reductase of strain K9, was cloned, and effectively expressed in E. coli. The association of the tmr2 gene with a typical mobile element ISPpu12 both existed in strain K9 and another MG-degrading strain MDB-1, which indicated that the tmr2 gene might be a horizontally transferable gene.
References
Alderman DJ (1985) Malachite green: a review. J Fish Dis 8:289–298
An SY, Min SK, Cha IH, Choi YL, Cho YS, Kim CH, Lee YC (2002) Decolorization of triphenylmethane and azo dyes by Citrobacter sp. Biotechnol Lett 24:1037–1040
Ayed L, Chaieb K, Cheref A, Bakhrouf A (2009) Biodegradation of triphenylmethane dye Malachite Green by Sphingomonas paucimobilis. World J Microbiol Biotechnol 25:705–711
Cha CJ, Doerge DR, Cerniglia CE (2001) Biotransformation of malachite green by the fungus Cunninghamella elegans. Appl Environ Microbiol 67:4358–4360
Culp SJ, Beland FA (1996) Malachite green: a toxicological review. Int J Toxicol 15:219–238
Daneshvar N, Ayazloo M, Khataee AR, Pourhassan M (2007) Biological decolorization of dye solution containing Malachite Green by microalgae Cosmarium sp. Bioresour Technol 98:1176–1182
Eichlerova I, Homolka L, Nerud F (2006) Synthetic dye decolorization capacity of white rot fungus Dichomitus squalens. Bioresour Technol 97:2153–2159
European Commission (2006) The rapid alert system for food and feed (RASFF) annual report 2005, Official publications of the European communities, Luxembourg
Fernandes C, Lalitha VS, Rao KVK (1991) Enhancing effect of malachite green on the development of hepatic pre-neoplastic lesions induced by N-nitrosodiethylamine in rats. Carcinogenesis 12:839–845
Hoffman GL, Meyer FP (1974) Parasites of freshwater fishes. a review of their control and treatment. TFH Publications, Neptune, New Jersey, p 224
Holt JG, Krieg NR, Sneath PHA, Staley JT, Williams ST (1994) Bergey’s manual of determinative bacteriology, 9th edn. Wilkins, USA
Jadhav JP, Govindwar SP (2006) Biotransformation of malachite green by Saccharomyces cerevisiae MTCC 463. Yeast 23:315–323
Jang MS, Lee YM, Kim CH, Lee JH, Kang DW, Kim SJ, Lee YC (2005) Triphenylmethane reductase from Citrobacter sp. Strain KCTC18061P: purification, characterization, gene cloning, and overexpression of a functional protein in Escherichia coli. Appl Environ Microbiol 71:7955–7960
Li LT, Hong Q, Yan X, Fang GH, Shinawar WA, Li SP (2009) Isolation of a malachite green-degrading Pseudomonas sp. MDB-1 strain and cloning of the tmr2 gene. Biodegradation 20:769–776
Littlefield NA, Blackwell BN, Hewitt CC, Gaylor DW (1985) Chronic toxicity and carcinogenicity studies of gentian violet in mice. Fundam. Appl Toxicol 5:902–912
Liu WX, Chao YP, Yang XQ, Bao HB, Qian SJ (2004) Biodecolorization of azo, anthraquinonic and triphenylmethane dyes by white-rot fungi and a laccase-secreting engineered strain. J Ind Microbiol Biotechnol 31:127–132
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 16:1215
Parshetti G, Kalme S, Saratale G, Govindwar S (2006) Biodegradation of Malachite Green by Kocuria rosea MTCC 1532. Acta Chim Slov 53:492–498
Rao KVK (1995) Inhibition of DNA synthesis in primary rat hepatocyte cultures by malachite green: a new liver tumor promoter. Toxicol Lett 81:107–113
Ren SZ, Guo J, Zeng GQ, Sun GP (2006) Decolorization of triphenylmethane, azo, and anthraquinone dyes by a newly isolated Aeromonas hydrophila strain. Appl Microbiol Biotechnol 72:1316–1321
Sani RK, Banerjee UC (1999) Decolorization of triphenylmethane dyes and textile and dye-stuff effluent by Kurthia sp. Enzyme Microb Technol 24:433–437
Schnick RA (1988) The impetus to register new therapeutics for aquaculture. Prog Fish-Cult 50:190–196
Shin KS, Kim CJ (1998) Decolorization of artificial dyes by peroxidase from the white-rot fungus, Pleurotus ostreatus. Biotechnol Lett 20:569–572
Sudova E, Machova J, Svobodova Z, Vesely T (2007) Negative effects of malachite green and possibilities of its replacement in the treatment of fish eggs and fish: a review. Vet Med 52:527–539
Tekere M, Mswaka AY, Zvauya R, Read JS (2001) Growth, dye degradation and ligninolytic activity studies on Zimbabwean white rot fungi. Enzyme Microb Technol 28:420–426
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Utkarsha S, Rhishikesh D, Jyoti J (2008) Biodegradation of triphenylmethane dye cotton blue by Penicillium ochrochloron MTCC 517. J Hazard Mater 157:472–479
Wamik A, Rajesh KS, Uttam CB (1998) Biodegradation of triphenylmethane dyes. Enzyme Microb Technol 22:185–191
Wang SM, He J, Cui ZL, Li SP (2007) Self-formed adaptor PCR: a simple and efficient method for chromosome walking. Appl Environ Microbiol 73:5048–5051
Weightman AJ, Topping AW, Hill KE, Lee LL, Sakai K, Slater JH, Thomas AW (2002) Transposition of DEH, a broad-host-range transposon flanked by ISPpu12, in Pseudomonas putida is associated with genomic rearrangements and dehalogenase gene silencing. J Bacteriol 184:6581–6591
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Acknowledgments
This work was supported by Natural Science Foundation of Jiangsu Province, China (BK2009312), Chinese National Programs for High Technology and Development (2007AA061101), The Opening Fund of State Key Laboratory of Soil and Sustainable Agriculture (Y052010025).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huan-Mei, Lian-Tai, L., Cai-Fang, Y. et al. Biodegradation of malachite green by strain Pseudomonas sp. K9 and cloning of the tmr2 gene associated with an ISPpu12 . World J Microbiol Biotechnol 27, 1323–1329 (2011). https://doi.org/10.1007/s11274-010-0580-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-010-0580-7