
Abstract
It is widely reported that coral reefs are suffering degradation from a combination of stresses. We have previously reported the use of genomic tools in the study of environmental impact assessment and hypothesize that monitoring the bacterial diversity associated with a coral would indicate changes in its health before visible damage occurs. This study analyzes the bacterial diversity associated with Porites coral collected from two sites in the Arabian Sea using culture independent techniques. Two clone libraries were constructed from the16S rDNA amplicons and selected individual clones were partially sequenced. Retrieved sequences were identified by BLAST analysis and indicate the presence of unidentified bacteria. Diversity index was calculated using Shannon–Wiener index. The bacterial diversity associated with coral sample 1 gives the index of species diversity, H 1 as 1.6287, divergence from eqiprobability as 45.708% and the evenness of the sample as 54.291. For coral sample 2, the values obtained were, H 1 as 1.97, divergence from eqiprobability as 15.11% and the evenness of the sample as 84.88. Sequences representing bacteria related to agricultural or industrial pollution and pathogenesis were also found. Coral samples collected near a lagoon area, showed a greater percentage of sequences representing bacteria related to human interventions, indicative of pollution.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The association between bacteria and corals is said to play an important role in coral health (Bourne and Munn 2005). Numerous investigators have examined the interactions between corals and microbes (Lewis and Price 1975; Ducklow and Mitchell 1979; Ritchie and Smith 1995; Richardson 1998; Dobretsov and Qian 2004; Bourne and Munn 2005). These studies have shown that there is a dynamic microbiota living on the surface and probably within the tissue of corals and in the surrounding waters. The bacterial communities associated with corals are found to be largely coral specific (Ritchie and Smith 2004). Shifts in bacterial community could affect the health of the coral. It has been demonstrated that the bacterial community of the whole coral colony was affected even when a very small part of the colony showed signs of disease (Pantos et al. 2003). Bacteria associated with the soft coral Dendronephthya sp. have been shown to contribute to the antifouling mechanisms of the soft-bodied organisms by producing compounds that inhibit bacterial growth and settlement of macrofoulers on the surface of their host (Dobretsov and Qian 2004). Understanding the microbial community is the key to understanding this unique ecosystem and ecosystem disturbances.
Water quality in costal areas around the world is changing in response to agricultural and industrial runoffs, and sewage discharge (Vitousek et al. 1997; Tilman et al. 2001; Fabricius 2005). Shifts in the normal microbiota have been observed even before visible signs of stress have recorded. This suggests that monitoring the microflora could be used as bioindicators of environmental change. Stress as a result of increased human impacts on costal regions may affect the microbial community, which in turn will have adverse effects on the health of corals. This study analyzes the bacterial diversity associated with the Porites coral, amongst the most abundant coral species in the world, and demonstrates the presence of bacteria not related to this marine ecosystem.
Despite rapid advances in microbial diversity studies, only 1% of the diversity is known (Pace 1997). Culture based techniques cannot reveal a large part of the bacterial population (Fuhrman and Campbell 1998). Advances in DNA technology has allowed researches to overcome this drawback and led to a detailed investigation into the diversity. PCR using universal 16S rRNA primers is the most commonly used tool. This study identifies the microbial population associated with Porites spp. using culture independent methods. Partial sequence data has revealed a large population of unidentified bacteria associated with the coral sample. The sample closer to human influence (lagoon area) also shows the presence of bacteria related to disease. Results also show the presence of sequences related to bacteria usually reported in degradation of pesticides and herbicides, suggesting pollution effects by water contamination due to agricultural land runoffs.
Materials and methods
Sample location and collection
Samples of Porites spp. were collected from two sites in the Arabian Sea, near the coast of India. Sample 1 was collected off Panaji, Goa, India at location Latitude 15.30°N, Longitude 73.55°E. Sample 2 was collected from lagoon area in Kavaratti island, Lakshadweep, Latitude 10.57°N : Longitude 72.64°E. All samples were collected by divers between 4 and 5 m in depth. Drill cores from the top portion were collected and immediately placed in a plastic bag. For each sample, drill cores were collected from five different polyps. At the surface, each sample was washed with filter-sterilized sea water and transported to the lab within 24 h. The coral samples were transferred into sterile 50 ml tubes and immersed in 25 ml phosphate buffered saline (8 g NaCl, 0.2 g KCl, 2.68 g Na2HPO4-7H2O, and 0.24 g KH2PO4 in 1 l of water). The tubes were then incubated in a water bath sonicator set at 60 sonics per mic (Branson 2210 Ultrasonic waterbath, USA) for 10 min to dislodge the bacteria adhered to the coral surface. Five-milliliter aliquots of slurry were collected into a centrifuge tubes and processed for preparation of DNA. One aliquot was preserved at −20°C.
DNA extraction
The bacterial population associated with the coral sample was analyzed by using unculturable techniques. PCR compatible DNA was made from 5 ml slurry, as described above. The slurry was centrifuged at 10,000 g for 30 min. and DNA was prepared as reported earlier (Kapley et al. 2000). The supernatant was discarded and pellet was resuspended in 25 μl 0.5 N NaOH and incubated for 30 min at room temperature. About 25 μl 1 M Tris, pH 7.5 was added to neutralize the suspension and volume made up to 500 μl with autoclaved deionized water (Elix 3, Millipore, Vienna, Austria). Extraction with an equal volume of phenol-chloroform mixture was followed by ethanol precipitation. The DNA pellet was resuspended in 50 μl 10 mM Tris pH 7.5. DNA extracted from all five replicates of each sample were pooled to create homogeneity and stored at −20°C until required. Five micro liter was used as template for PCR.
Construction and analysis of 16S rDNA library
16S rDNA gene was amplified using universal eubacterial primers as reported earlier (Narde et al. 2004). A 1,466 bp product was amplified using forward primer 5′-AGAGTTTGATCMTGGCTCAG-3′ and reverse primer 5′-TACGGYTACCTTGTT ACGACTT-3′. The reaction mixture comprised of 100 pM primers, 200 μM each dNTP, 3 mM MgCl2, buffer and 2.5 units of Amplitaq DNA polymerase (Perkin Elmer, Turku, Finland) in a total volume of 50 μl. Thermocycling conditions were 30 cycles of, denaturation at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1 min. For each sample, ten individual reactions were set and analyzed on 1% agarose gel. 16S rDNA amplification products were pooled and then were extracted from the gel using QIA gel extraction kit (Qiagen, Hilden, Germany) and cloned using the TA Cloning Kit (Qiagen) according to the instructions of the manufacturer. Transformants were selected on Luria-Bertani (LB) agar plates containing kanamyxin, X-gal and IPTG following the manufacturer’s instructions. White colonies were picked and plasmid prep was carried out using standard alkaline lysis protocol. Plasmid DNA was digested with EcoRI to check the presence of insert. Universal 16S primers were used to amplify the insert from the plasmid DNA. The resultant amplicon was gel purified (Qiagen gel purification kit) and amplified ribosomal DNA restriction analysis (ARDRA) was carried out using HaeIII enzyme. Clones showing different band patterns were selected for sequencing of the 16S rDNA insert. Sequencing was done commercially (Bangalore Genei, Bangalore, India). Partial sequencing was carried out from the 5′ end using T7 primer. The sequence data (450–750 bp) was analyzed by BLAST to identify the nearest previously described bacteria. Sequences were deposited in GenBank (http://www.ncbi.nlm.nih.gov/).
Phylogenetic analysis
Phylogenetic Trees were constructed using the BOOTSTRAP Tree method from Clustal × software, originally developed by Thompson et al. (1997). The method involves aligning sequences using the Neighbor Joining (NJ) method. First, distances are calculated (percent divergence) between all pairs of sequence from a multiple alignment and then, the NJ method is applied to the distance matrix. The tree is created using the BOOTSTRAP N-J TREE method. It uses a method for deriving confidence values for the groupings in a tree. It involves making N random samples of sites from the alignment, drawing N trees (one from each sample) and counting how many times each grouping from the original tree occurs in the sample trees. About 1,000 iterations were carried out for each analysis.
Diversity index
The Shannon–Wiener diversity index (H) was used to characterize species diversity in the sample (Shannon 1948). Shannon’s index accounts for both abundance and evenness of the species present, and is given by the following equation:
where H = Information content of sample, Index of species diversity, or Degree of Uncertainty, s = Number of species, and p i = Proportion of total sample belonging to ith species.
Measures of evenness
The maximum Shanon–Wiener index for a given number of species is calculated by:
The minimum Shannon–Wiener index for a given data set can be calculated by:
where S is the number of categories or species and N is the total number of observations.
The evenness of the sample is calculated using the following two equations:
The divergence from eqiprobability is defined as:
.
Results
Bacteria associated with coral samples
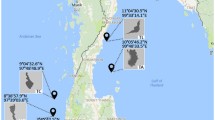
Two sampling sites were chosen for collection of the Porites spp. on the west coast of India as shown in Fig. 1. Sample 2 was collected from the lagoon area so as to represent a site, which would have possible effects of human interventions. 16S rDNA library was made and clones were screened by ARDRA (data not shown) and selected clones were sequenced. The sequence data was analyzed by BLAST and the nearest match from GenBank data was reported. Sequences were deposited in the GenBank and accession numbers are noted in Figs. 2 and 4, respectively. Sample 1 showed 58.69% sequences representing uncultured bacteria. About 28% of the clones showed sequence identity with Caulobacter sp. The genus Caulobacter is composed of prosthecate bacteria often specialized for oligotrophic environments (Abraham et al. 1999). The uncultured caulobacter identified here, have been segregated into two groups as can be seen in the phylogenetic tree (Fig. 2). However, when the sequences were individually aligned with control Caulobacter sequences from GenBank (culturable and uncultured clones), using the MegAlign program of DNASTAR (Laser Gene software), they appear to originate from the same branch of the tree. However, they are further subdivided into two branches as shown in Fig. 3. Since the sequences represent unculturable population, it is quite possible that some of them could belong to a taxonomy closely related to Caulobacter. Bacterial sequences that show >90% identity to sequences reported in human diseases have also been observed. For e.g., uncultured Veilonella clone, HKT9, shows 99.6 sequence identity, gap length 0, (using the Wilbur–Lipman alignment algorithms are available for pairwise alignments, DNASTAR) with a Veilonella oral clone (GenBank accession no. AF287782) observed in a study of human subgingival plaque (Paster et al. 2001). Clone HKT27, uncultured Dolosigranulum shows 96.9% similarity index with gap length of 6 to Dolosigranulum pigrum from human infections (GenBank accession no. X70907). A large number of clones showed sequence identity to an unidentified culturable bacteria that was isolate from activated biomass of an effluent treatment plant treating wastewater from industry (isolate HPC34, accession no. AY803979).

Map of sample location site in the Arabian Sea, off the west coast of India. Sample 1 was collected off Panaji, Goa, India at location Latitude 15.30°N, Longitude 73.55°E. Sample 2 was collected from lagoon area in Kavaratti island, Lakshadweep, Latitude 10.57°N : Longitude 72.64°E

16S rDNA phylogenetic tree of sequences analyzed from clones of 16S rDNA library of coral sample 1, collected off Goa coast, location Latitude 15.30°N, Longitude 73.55°E. The tree was constructed by bootstrapping (1,000 iterations). The accession numbers of the clones reported in this study are given in square brackets. Control sequences downloaded from GenBank and used in tree construction are indicated in bold typeface with round brackets

Phylogenetic tree showing multiple sequence alignment of Caulobacter clones reported in this study along with culturable and unculturable Caulobacter species reported in GenBank. The tree was constructed using the MegAlign program of DNASTAR (LaserGene software, Madison, WI, USA). The clones reported in this study are prefixed by HKT, while sequences downloaded from GenBank are prefixed with accession number followed by the identification
Coral sample 2, yielded 48.38% uncultured bacteria. Most sequences show identity with bacterial diversity studied in deep sea sites near Japan (Takami et al. 1999). Clones from coral sample 2 showed sequence identity to a large number of bacteria that are reported in degradation of pesticides, herbicides and insecticides (Martin et al. 1999; Kumar and Philip 2006). Clones HKT72, 78 and 91 were identified by BLAST as uncultured Chryseobacterium reported in diseased aquatic animals and hospital wastes (Bernardet et al. 2005; Akay et al. 2006). The phylogenetic tree of bacteria associated with coral sample 2 is shown in Fig. 4. In this tree, clone HKT67, HKT63, and HKT62, identified as Acinetobacter, do not appear along with other Acinetobacter clone sequences. Hence, we compared the sequence data to culturable and unculturable Acinetobacter reported in GenBank. A phylogenetic tree was constructed using the MegAlign program of DNASTAR and is shown in Fig. 5. As can be seen in the figure, all Acinetobacter clones align together. However, they could be more closely related to the unidentified clones and hence the disparity between Acinetobacter clones in the bootstrap tree (Fig. 4). For e.g., clone HKT62 (uncultured Acinetobacter) shows 96.9 similarity index with HKT59 (uncultured bacterium clone) and 98.3 similarity index with clone HKT79 (uncultured bacterium clone).

16S rDNA phylogenetic tree of sequences analyzed from clones of 16S rDNA library of coral sample 2, collected from lagoon area, Kavaratti island, Lakshadweep, Latitude 10.57°N : Longitude 72.64°E. The tree was constructed by bootstrapping (1,000 iterations). The accession numbers of the clones reported in this study are given in square brackets. Control sequences downloaded from GenBank and used in tree construction are indicated in bold typeface with round brackets

Phylogenetic tree showing multiple sequence alignment of Acinetobacter clones reported in this study along with culturable and unculturable Acinetobacter species reported in GenBank. The tree was constructed using the MegAlign program of DNASTAR (LaserGene software). The clones reported in this study are prefixed by HKT, while sequences downloaded from GenBank are prefixed with accession number followed by the identification
Diversity index
A diversity index is a mathematical measure of species diversity in a community. Shannon’s index accounts for both abundance and evenness of the species present. The bacterial diversity associated with coral sample 1 shows index of species diversity, H 1 as 1.6287 and H 1max as 3. The divergence from eqiprobability (D 1) was calculated to be 1.371 (D 1% = 45.708) and the evenness of the sample was 54.291. For coral sample 2, the values obtained were H 1 as 1.971 and H 1max as 2.32. The divergence from eqiprobability was calculated to be 0.351 (D 1% = 15.11) and the evenness of the sample was 84.88. A low D 1 value means H 1 is close to H 1max, or, the system is nearly in a state of equiprobability; there is a high degree of diversity present. Conversely, a high D 1 value means that H 1 is small relative to H 1max, that is, the system has diverged substantially from equiprobability and is not very diverse. This indicates that bacterial diversity associated with coral sample 2, where D = 0.351, has a higher degree of diversity. Diversity indices are useful, but only give an approximate idea of microbial diversity.
Discussion
Pollution in the environment is causing serious concerns globally. On analysis of the bacterial community associated with coral samples, we came across unexpected sequence data, which suggests traces of pollution in the marine environment. Sequence data of the 16S rDNA libraries of both samples demonstrated sequences with >99% identity with sequences of bacteria related to degradation of xenobiotics and possibly human infections. This result was not unexpected from the sampling site near the lagoon area as it was located close to human population. However, sequence identity results suggests the presence of bacteria related to land run-offs and hospital wastes in both coral samples. The diversity index of sample 2 was found to be higher. This is probably due to the presence of clones that show sequence identity to bacteria not related to the marine ecosystem.
There are various reports on the assessment of microbial diversity from marine environments like corals, estuarine waters and brackish sediments (Rohwer et al. 2001; Hewson and Fuhrman 2006; Abbondanzi et al. 2005). However, to our knowledge, this is the first report of bacterial diversity associated with the Porites spp. in the Arabian Sea. Contamination of sea water is being reported in the marine ecosystem due to xenobiotics used in pharmaceuticals and pesticides and from municipal sewage (Fent 1989; Aguilar and Borrell 2005). These alarming reports do not bode well for the health of the marine ecosystem and would endanger the health of the coral reefs and lead to degradation of this ecosystem We have previously reported the use of genomic tools to analyze environment impact assessment (Purohit et al. 2003). Future studies in this area would be the analysis of the bacterial diversity data to predict the degradation of the coral reefs before visible damage sets in. this would be a challenging area to succeed in preservation of the marine ecosystem. It could be argued that culturable bacterial profile would also help, but practical difficulties in sample transportation and survival of bacteria would increase the error in the study. This study provides an insight into the bacteria diversity associated with corals.
Conclusion
This study reveals the association of bacteria and coral. Clone sequence data showing strong identity with bacteria associated with human activity like agriculture and health have been observed in both samples. This study suggests the pollution in marine ecology, but to confirm these observations, the representing bacteria need to be cultured and identity confirmed by sequence analysis. Further studies also need to include the analysis of water surrounding the coral. It is recommended that policy controls need to be brought in to make sure that only treated waters are allowed to enter the marine ecosystem. In the long-term this may have severe consequences for the functioning of coastal ecosystems such as coral reefs. This is obviously an area for future research, since herbicides are likely to reach the coastal zone in combination with other stressors such as heavy metals, nutrients, sediments etc., and might thus lead to synergistic effects.
References
Abbondanzi F, Campisi T, Focanti M et al (2005) Assessing degradation capability of aerobic indigenous microflora in PAH-contaminated brackish sediments. Mar Environ Res 59:419–434
Abraham WR, Strompl C, Meyer H et al (1999) Phylogeny and polyphasic taxonomy of Caulobacter species Proposal of Maricaulis gen nov with Maricaulis maris (Poindexter) comb nov as the type species, and emended description of the genera Brevundimonas and Caulobacter. Int J Syst Bacteriol 49:1053–73
Aguilar A, Borrell A (2005) DDT and PCB reduction in the western Mediterranean from 1987 to 2002, as shown by levels in striped dolphins (Stenella coeruleoalba). Mar Environ Res 59:391–404
Akay M, Gunduz E, Gulbas Z (2006) Catheter-related bacteremia due to chryseobacterium indologenes in a bone marrow transplant recipient. Bone Marrow Transpl PMID:16415896 37:435–436
Bernardet JF, Vancanneyt M, Matte-Tailliez O et al (2005) Polyphasic study of Chryseobacterium strains isolated from diseased aquatic animals. Syst Appl Microbiol 28:640–660
Bourne DG, Munn CB (2005) Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Environ Microbiol 7:1162–1174
Dobretsov S, Qian P-Y (2004) The role of epibiotic bacteria from the surface of the soft coral Dendronephthya sp in the inhibition of larval settlement. J Exp Mar Biol Ecol 299:35–50
Ducklow HW, Mitchell R (1979) Bacterial populations and adaptations in the mucus layers on living corals. Limnol Oceanogr 24:715–725
Fabricius KE (2005) Effects of terrestrial runoff on the ecology of corals and coral reefs: review and synthesis. Mar Pollut Bull 50:125–146
Fent K (1989) Organotin speciation in municipal wastewater and sewage sludge: Ecotoxicological consequences. Mar Environ Res 28:477–483
Fuhrman JA, Campbell L (1998) Microbial microdiversity. Nature 393:410–411
Hewson I, Fuhrman JA (2006) Improved strategy for comparing microbial assemblage fingerprints. Microbial Ecol PMID: 16437287 5:147–153
Kapley A, Lampel K, Purohit HJ (2000) Development of duplex PCR for Salmonella and Vibrio. World J Microbiol Biotechnol 16:457–58
Kumar M, Philip L (2006) Enrichment and isolation of a mixed bacterial culture for complete mineralization of endosulfan. J Environ Sci Heal B 41:81–96
Lewis JB, Price WS (1975) Feeding mechanisms and feeding strategies of Atlantic reef corals. J Zool 176:527–544
Martin M, Mengs G, Allende JL et al (1999) Characterization of two novel propachlor degradation pathways in two species of soil bacteria. Appl Environ Microb 65:802–806
Narde G, Kapley A, Purohit HJ (2004) Isolation and characterization of Citrobacter strain HPC 255 for broad range substrate specificity for chlorophenol. Curr Microbiol 48:419–423
Pace NR (1997) A molecular view of microbial diversity and the biosphere. Science 276:734–740
Pantos O, Cooney RP, Le Tissier MDA (2003) The bacterial ecology of plaque-like disease affecting the Caribbean coral Montastrea annularis. Environ Microbiol 5:370–382
Paster BJ, Boches SK, Galvin JL et al (2001) Bacterial diversity in human subgingival plaque. J Bacteriol 183:3770–3783
Purohit HJ, Raje DV, Kapley A et al (2003) Genomics tools in environmental impact assessment. Environ Sci Technol 37:356A–363A
Richardson LL (1998) Coral diseases: what is really known? Trends Ecol Evol 13:438–443
Ritchie KB, Smith GW (1995) Preferential carbon utilization by surface bacterial communities from water mass, normal and white band diseased Acropora cervicornis. Mol Mar Biol Biotech 4:345–354
Ritchie KB, Smith GW (2004) Coral health and disease. Springer-Verlag, New York
Rohwer F, Breitbart M, Jara J et al (2001) Diversity of bacteria associated with the Caribbean coral Montastraea franksi. Coral Reefs 20:85–91
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423
Takami H, Kobata K, Nagahama T et al (1999) Biodiversity in deep-sea sites located near the south part of Japan. Extremophiles 3:97–102
Tilman D, Fargione J, Wolff B et al (2001) Forecasting agriculturally driven global environmental change. Science 292:281–284
Thompson JD, Gibson TJ, Plewniak F et al (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Vitousek PM, Aber JD, Howarth RW (1997) Human alteration of the global nitrogen cycle: sources and consequences. Ecol Appl 7:737–750
Acknowledgments
This study was supported by a grant from the Council of Scientific And Industrial Research, India, project SMMOOO2.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kapley, A., Siddiqui, S., Misra, K. et al. Preliminary analysis of bacterial diversity associated with the Porites coral from the Arabian sea. World J Microbiol Biotechnol 23, 923–930 (2007). https://doi.org/10.1007/s11274-006-9315-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-006-9315-1