
Abstract
After mine closure in the 1980s and subsequent shutdown of the dewatering system, groundwater rebound led to drainage outflow from the Casargiu gallery (Montevecchio mine, SW Sardinia, Italy) beginning in 1997. Mine drainage had pH 6.0 and dissolved concentrations of sulfate (5000 mg/L) and metals (e.g., 1000 mg/L Zn, 230 mg/L Fe, 150 mg/L Mn) much higher than those previously measured in groundwater under dewatering conditions. As compared with the first stages of rebound at Casargiu, a very high contamination level still persists after more than 15 years of flushing. Mine drainage (20–70 L/s; pH 6.0 ± 0.2; Zn-Mg-Ca-SO4 composition) flowed into the Rio Irvi. Abundant precipitation of amorphous Fe(III)-(oxy)hydroxides occurred. Moreover, sulfate-bearing green rust was observed to flocculate in the reach of the Rio Irvi where pH was still circumneutral. Water sampling along this stream for about 6 km almost to its mouth in the Mediterranean Sea showed a pH decrease from 6.0 to 4.0 and a significant removal of Fe (46 %) and As (96 %), while sulfate, Zn, Mn, Co, Ni, and Cd showed small variations downstream. Lead was initially adsorbed onto Fe(III)-(oxy)hydroxides, then desorbed as pH dropped below 5. The estimated amount of dissolved metals discharged into the Mediterranean Sea is significant (e.g., 900 kg/day Zn, 1.4 kg/day Cd, 5 kg/day Ni). In particular, a conservative estimation of the amount of Zn discharged to the sea is about 330 ton/year, which would correspond to 1.4 % of the global annual flux of dissolved Zn from uncontaminated rivers to the oceans.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
A problem often associated with mine closure is the shutdown of pumping for dewatering of underground mining works (Younger 2000; Adams and Younger 2001; Bain et al. 2001; Razowska 2001; Cidu et al. 2007; Gandy and Younger 2007). The consequent rebound of the water table causes flooding of galleries and interaction of groundwater with exposed mineralization and mining wastes used to fill galleries and voids (Rosner 1998; Royle 2007). The contaminated groundwater may reach the surface through shafts and/or galleries or previously active springs, with discharges that have detrimental effects on the hydrochemistry and biological integrity of receiving waters (Wood et al. 1999; Younger 2001; Gzyl and Banks 2007). In fact, these water discharges generally have high total dissolved solids (often a sulfate-dominated composition), are heavily contaminated with toxic elements, and may have either an acidic pH (e.g., Geldenhuis and Bell 1997; Gray 1997) or a circumneutral pH (e.g., Banks et al. 1997; Younger 2001; Iribar 2004; Cravotta 2007) depending on the acid-neutralizing capacity of gangue minerals and host rocks. A long-term evolution in the quality of discharged water is to be expected, as reported by Wood et al. (1999) for abandoned coal mines in Scotland. They found that mine water pollution is most severe in the first few decades after a discharge begins (the “first flush”), and that even the largest systems settle down to a lower level of pollution (particularly in terms of iron concentration) within 40 years. With regard to mine water remediation, Wood et al. (1999) suggested active treatment of discharges for the first decade or two, followed by long-term passive treatment after pollutant concentrations asymptotically approach lower levels.
Sardinia is an Italian region with mining activities dating back to pre-Roman times. Peaks in Pb-Zn production were reached in the 1950s–1960s. The decline of the mining industry, due to the fall in value of base metals and the increase of labor costs, led to the closure of base-metal mines from 1970 to 1990. The potential adverse effects of mine closure were not given proper consideration, especially the risk to human health due to the proximity of mining sites to water resources. The chemical contamination of aquatic systems at abandoned mining sites requires investigation strategies capable of describing sources and pathways of contaminants and containment strategies devoted to limit the dispersion of toxic components (RAS 2003; WHO 2006).
This paper focuses on assessing the environmental effects of mine drainage outflow from the Casargiu gallery (Montevecchio mine, SW Sardinia, Italy) on the aquatic system (in particular the Rio Irvi) and on metal transport to the Mediterranean Sea. Temporal variations of mine drainage chemistry and distribution of dissolved sulfate and metals along the Rio Irvi over a distance of about 6 km are presented and discussed with the aim of understanding the geochemical-mineralogical processes taking place and estimating the metal load discharged into the sea.
2 Study Area
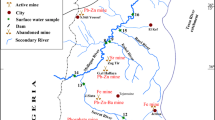
The Arburese mining district (SW Sardinia, Fig. 1) is mainly composed of the mine workings of Montevecchio and Ingurtosu. Casargiu is one of the mines belonging to the Montevecchio mining system. The Pb-Zn sulfide veins are hosted in Palaeozoic silicate-dominant rocks. These ore deposits have variable compositions both in sulfide and gangue minerals; galena and sphalerite are by far the most abundant sulfides, with minor amounts of chalcopyrite, pyrite, and marcasite. The gangue is comprised of quartz, barite, siderite, ankerite, and minor calcite and fluorite. In general, galena-rich veins are associated with a quartz-barite gangue, while sphalerite-rich veins have greater proportions of carbonate-rich gangue. Operationally, Casargiu is considered to be the westernmost extension of the Montevecchio mine, but geologically, it is actually an eastern offshoot of the Brassey vein of the Ingurtosu mine (Fig. 1). Mineralization at Casargiu mainly consists of sphalerite with an ankerite-siderite gangue (Biddau et al. 2001, and references therein; Concas et al. 2006).

Schematic map of the Arburese mining district (black square in the inset with Sardinia) showing the location of the Casargiu mine
Ore exploitation in the Montevecchio area extended in a system of interconnected galleries at depths down to 600 m below ground level. In particular, at the Casargiu mine, exploitation reached 180 m below the elevation of the Casargiu gallery, whose entry is 158 m above sea level. To keep the galleries dry, a total flow in the range of 55 to 70 L/s of water was pumped out of the mines of the Arburese mining district, including Casargiu. The mine closure in the 1980s led to the shutdown of pumping systems, thereafter drainage flowing out of the Casargiu gallery was observed beginning in 1997 (Caboi et al. 1999; Cidu and Fanfani 2002). The Casargiu drainage flows into the Rio Irvi that after about 6 km merges with the Rio Piscinas, which in turn flows into the Mediterranean Sea after about 2 km (Fig. 1).
The hydrological basin of the Rio Irvi has an area of about 15.4 km2 and a length of about 11 km. The Donegani and Zerbino water reservoirs (308,000 m3), located about 2 km upstream of the Casargiu mine, reduce the flow of the Rio Irvi to less than 10 L/s, making the outflow from the Casargiu gallery (20–70 L/s) the main water contribution to the Rio Irvi throughout the year.
Figure 1 shows the location of mines, flotation plants, and tailings. The tailings initially retained by dams in impoundments were periodically discharged into the local streams. At present, the dumped wastes are deeply eroded and most of the material has been transported and deposited on the stream banks (Caboi et al. 1999).
Climatic conditions in the region are characterized by periods of heat and drought, usually extending from May to September, interrupted by relatively short rainy periods, with occasional heavy rain events. Mean rainfall, calculated on pluviometric data of the Montevecchio rain gauge station for the 1922–2010 period (RAS 2013), is 735 mm/year, with a mean of 74 rainy days per year. July and August are the driest months, while November and December are the wettest months. The driest year was 1995 with a rainfall of just 376 mm and 41 rainy days, while 2010 was the wettest year (1215 mm, 111 rainy days). Compared to the mean rainfall of the 1922–2010 period, the last 15 years have been wetter, with 1154 mm in 1996, 1065 mm in 2004, and 973 mm in 2009. The mean rainfall of the 1995–2010 period is 824 mm, with a mean of 77 rainy days per year. The mean annual temperature from 1922 to 2010 is 15 °C.
3 Sampling and Methods
3.1 Waters
Sampling and analyses of waters draining the Casargiu mine area have been carried out under different seasonal conditions from 1997 to the present. The location of sampling sites is shown in Fig. 1. Water samples were labeled as CAS#. The water flowing out of the Casargiu gallery (CAS1) was collected during 23 surveys carried out from 1997 to 2012. Results of these surveys will be used to evaluate temporal variations in chemistry of the Casargiu discharge.
In June 2009, we carried out the most complete sampling of the Rio Irvi catchment, including the Casargiu discharge. The Rio Irvi was sampled at eight sites (CAS2 to CAS9) down to about 6 km downstream of the Casargiu gallery (CAS1). The last water sampling site (CAS9) was located 1 km downstream of the confluence with the Rio Piscinas and 1 km upstream of the mouth (Fig. 1). The Rio Irvi was mainly fed by the Casargiu drainage (about 50 L/s). The small tributaries of the Rio Irvi were dry, as was the Rio Piscinas at the confluence with the Rio Irvi (Fig. 1). This observation allows us to rule out lateral surface inputs of water to the Rio Irvi, with the exception of the Casargiu discharge. Results of the 2009 survey will be used to evaluate the dispersion of contaminants and estimate the metal load to the Mediterranean Sea.
The same methods were used for all the surveys, unless otherwise specified. At each sampling site, the pH, redox potential (Eh, Orion Pt electrode), dissolved oxygen (YSI Incorporated Ohio, Model 50B dissolved oxygen meter), temperature, and alkalinity were measured; all water samples were filtered through 0.4-μm pore-size Nuclepore polycarbonate filters into pre-cleaned high-density polyethylene bottles. Filtered aliquots were acidified on site with suprapure-grade HNO3 for metal analyses by quadrupole inductively coupled plasma–mass spectrometry (ICP-MS) and major cations by inductively coupled plasma–optical emission spectrometry (ICP-OES). Anions were determined by ion chromatography (IC) on a filtered (0.4 μm), unacidified aliquot.
In the June 2009 survey, dissolved ferrous iron was determined on site (Merck SQ118 photometer, Merck 1.00796.001 kit); aqueous speciation of Fe was clearly dominated by ferrous iron, but the method was not sensitive enough to determine the low concentrations of ferric iron (FeIII = Fetotal − FeII) occurring in all samples.
During the sampling campaign performed in June 2009, water samples were filtered in situ using both 0.4- and 0.01-μm filters. No significant differences between chemical analyses from the two filter sizes were observed; thus, the dissolved concentrations determined in the aqueous fraction <0.4 μm were used in this paper.
Speciation-solubility calculations were performed using the computer program PHREEQC Interactive (version 3.1.2.8538 released on March 3, 2014; Parkhurst and Appelo 1999) with the included thermodynamic database “wateq4f.dat”. The ionic charge balance calculated with PHREEQC was in the range of 3 to 0.7 %, with the exception of CAS8 (7 %).
3.2 Solid Materials
In June 2009, solid precipitates were collected at the Casargiu outflow and from the streambed at each sampling site. Solid samples were labeled as CAS#s. It was very difficult to sample green rust from the streambed. Thus, the flocculant retained on a 0.45-μm filter upon water filtration at the CAS4 sampling site, where green rust precipitation appeared to predominate over Fe(III)-(oxy)hydroxides precipitation, was used. Solid samples were left to dry at room temperature and later ground in an agate mortar. Mineralogical analyses by powder X-ray diffraction (XRD) were performed using an X’pert Pro diffractometer (Panalytical) with θ-θ geometry, Cu-Kα1 wavelength radiation (λ = 1.54060 Å), and X’Celerator detector, operating at 40 kV and 40 mA. Patterns were collected from 3° to 75° 2θ with a step size of 0.02° 2θ.
Chemical analysis of solid precipitates was performed using about 0.1 g of each sample. A high-purity mixture of 3 mL of Milli-Q water, 1 mL of H2O2, 12 mL of aqua regia (3HCl:1HNO3), and 3 mL of HF was added to the solid samples in microwave vessels. Samples were placed in the microwave carousel together with a blank solution prepared with the same mixture. After microwave digestion, the vessel caps were removed and washed with small volumes of Milli-Q water; the solutions were transferred into Teflon beakers and heated on a hotplate (∼4 h at 150 °C). The solutions were then evaporated close to dryness; 3 mL of concentrated HNO3 was added, and this process was repeated three times. The final solutions were filtered (0.4 μm) and made up to 50 mL of final volume using Milli-Q water. Elements were determined by ICP-OES and ICP-MS.
4 Results and Discussion
4.1 Temporal Variations in Chemistry of the Casargiu Drainage
In 1973, under active mining at Casargiu (Biddau 1978), the pH of the water pumped out of the mine was near-neutral (pH = 7.5), but concentrations of sulfate and metals were already high (Table 1). However, the first outflow from the Casargiu gallery in 1997 (i.e., after mine closure) had a lower pH (pH = 6.0) and dissolved concentration of sulfate 3.5 times higher, as well as dissolved metals (especially Zn, Mn, and Fe) 1 or 2 orders of magnitude higher than those observed in groundwater under dewatering conditions (Table 1). These dramatic variations can be explained taking into account that the underground workings were kept dry during exploitation, and rebound of the water table after the shutdown of pumping systems led to a gradual flooding of the galleries. This flooding allowed the contact of groundwater with sulfide minerals (especially sphalerite), promoting their oxidation, and the dissolution of secondary minerals and soluble efflorescent salts that normally form in large amounts inside the mine galleries where high humidity conditions occur. Also, mine wastes and flotation tailings were used to refill some of the underground workings; the small grain size of these materials has certainly contributed to the extent and longevity of the water-sulfide interaction processes. The role of secondary minerals in affecting the geochemistry of water in mine environments is well known (e.g., Frau 2000). In the case of Casargiu, this role is especially evident when the concentrations of Zn and Cd and their ratio before and after the mine closure are taken into account (Table 1), and considering that Zn and Cd exhibit very similar geochemical behaviors in aqueous solution. During exploitation, the Zn/Cd ratio in groundwater pumped out (Zn/Cd = 58, Table 1) most likely reflected the Zn/Cd ratio in sphalerite (Zn/Cd = 67, value derived from analyses of sphalerite from Montevecchio; Palache et al. 1944), but this ratio is not conserved in common secondary Zn minerals, such as goslarite ZnSO4·7H2O, bianchite (Zn,Fe2+)SO4·6H2O, smithsonite ZnCO3, and hemimorphite Zn4Si2O7(OH)2·H2O, in which Cd does not enter or enters in much lower concentrations as compared to sphalerite (Palache et al. 1951). Therefore, after mine closure and groundwater rebound, the dissolution of these secondary Zn minerals produced a strong increase in dissolved Zn, while Cd remained almost constant (Zn/Cd = 500, Table 1).
Results of hydrogeochemical surveys carried out during the period 1997–2012 can be summarized as follows. The flow from the Casargiu gallery varied from 20 to 70 L/s (not shown) depending on rain infiltration, although no immediate correlation between rainfall and flow was observed and a mean time lag of about 3 months was estimated, providing evidence of a large hydrogeological system and/or slow movement (i.e., long residence time) of groundwater before emerging from the Casargiu gallery. Values of pH (6.0 ± 0.2) and Eh (0.28 ± 0.05 V) showed small variations over time (data not shown). The acidity produced by the oxidation of sulfide minerals (e.g., pyrite, Fe-bearing sphalerite) has been buffered by the occurrence of ankerite-siderite gangue in the Casargiu ore. Figure 2 shows temporal variations in total dissolved solids (TDS) and dissolved components in the CAS1 water. As compared with values recorded in the first stages of outflow at Casargiu, a significant decrease in TDS (Fig. 2a), sulfate and Zn (Fig. 2b), Ni and Co (Fig. 2c), and Pb (Fig. 2d) was observed with time, while Fe and Mn (Fig. 2b), Cd (Fig. 2c), and As (Fig. 2d) showed minor variations. The lowest values in sulfate and metals were generally observed during times with the highest flows. However, an estimation of the flow was not always possible because, in some periods, the mine drainage was not contained within the constructed channel and drainage water was spread across the surface near the channel. Therefore, temporal variations in concentrations of dissolved components in the Casargiu water discharge are not easily correlated with its flow, as well as with seasonal changes in rainfall. A very high contamination level still persists in the Casargiu drainage water after more than 15 years of flushing from the first outflow. Even though concentrations of various metals have decreased somewhat, they remain well above environmentally significant toxicity levels.

a–d Temporal changes in water chemistry at the Casargiu mine. In 1995, groundwater was sampled at depth from the shaft and its composition was still influenced by dewatering. The first outflow of groundwater from the Casargiu gallery occurred in 1997
4.2 Chemistry Changes Along the Rio Irvi
Tables 2 and 3 report the water chemistry along the Rio Irvi sampled for about 6 km downstream in June 2009. Waters from the Casargiu gallery and the Rio Irvi are characterized by a Zn-Mg-Ca-SO4 composition. In addition to the elements shown in Tables 2 and 3, we also determined Ag, B, Ba, Be, F, Li, Rb, and Tl (data not shown); these elements exhibited no significant variations along the stream and thus provided no insight into in-stream geochemical processes. Also Bi, Cr, Cu, Hg, Te, U, and V were determined, but concentrations were below the corresponding detection limits (Bi and U < 0.5 μg/L; Hg and Te < 1 μg/L; Cr, Cu, and V < 10 μg/L). The flow of the Casargiu drainage in June 2009 was about 50 L/s. Inputs to the Rio Irvi from tributaries were considered negligible because they were dry, although groundwater inputs cannot be excluded. The reach of the Rio Irvi extending upstream of the Casargiu discharge was uncontaminated (not shown).
Figure 3 shows the variation of pH and Eh with distance from Casargiu. A non-linear pH decrease (from 5.9 to 4.0) coupled with an increasing mirror trend of redox potential (Eh from 286 to 524 mV) can be observed. As reported in Table 1, the pH of the first outflow from the Casargiu gallery was significantly lower than the pH of groundwater pumped out during mining activity. In June 2009, the pH measured at CAS1 was 5.9; this pH value might be explained by the incomplete oxidation of sulfides and consequent buffering of acidity by the carbonate gangue, according to the following example reactions involving pyrite and siderite:

Variation of pH and Eh in stream water along the Rio Irvi sampled in June 2009
It is known that the pH of groundwater in a tailings impoundment in the presence of siderite is buffered at a value of about 6, and dissolved iron is present as Fe2+ owing to the low diffusivity of oxygen at depth that prevents Fe2+ from rapid oxidation to Fe3+ and also controls the Eh at sub-oxic values (Ptacek and Blowes 1994; Bain et al. 2000). Similar processes probably occur at Casargiu, where the use of flotation tailings to refill the underground workings might contribute to keep sub-oxic conditions because fine-grained materials, especially when wet, inhibit the diffusion of atmospheric oxygen. In fact, at CAS1, both measured and calculated aqueous speciation of Fe was dominated by Fe(II) (see Section 4.4) and dissolved oxygen (DO) was quite low (≤2 mg/L).
As the groundwater flowing out from the Casargiu gallery comes in contact with the oxygen of the atmosphere (DO = 7–8 mg/L at CAS2 and downstream), ferrous iron oxidizes to ferric iron and a typical ochreous precipitate is observed, according to the following reactions where a generic amorphous Fe(III)-hydroxide is taken as a possible precipitating phase:
Reaction (3) consumes H+, while reaction (4) produces H+. An increase of pH from 5.9 to 6.4 coupled with a slight decrease of Eh from 286 to 259 mV can be observed in the CAS1–CAS4 reach (Fig. 3). This result is probably due to an initially faster rate of reaction (3) compared to reaction (4), then production of H+ exceeds its consumption, pH decreases down to 4, and Eh increases up to 524 mV at CAS9. All along the Rio Irvi, ochreous precipitates cover the streambed, indicating a continuous formation of Fe(III)-(oxy)hydroxide; however, a peculiar flocculation of a green phase, which will be discussed in Section 4.3, especially occurs in the CAS3–CAS5 reach.
Figure 4 shows water chemistry changes along the Rio Irvi. With regard to major components, small negative percent variations of Ca (−5 %), Mg (−3 %) (Fig. 4a), SO4 (−7 %), Zn (−6 %), and Mn (−4 %) (Fig. 4b) occur from the Casargiu outflow to the last sampling point at a distance of 6 km (the percent variation is calculated as follows: 100 * (C CAS9 − C CAS1)/C CAS1, where C is the concentration in milligrams per liter). Alkalinity diminishes down to zero with pH (Fig. 4a), while dissolved Fe exhibits a strong decrease (−46 %, Fig. 4b) due to precipitation. Among minor and trace elements, negative variations of Ni (−9 %), Cd (−15 %), and Co (−11 %) might be due partly to co-precipitation/adsorption with Fe(III)-(oxy)hydroxide, while Pb exhibits a distribution consistent with adsorption/desorption onto/from Fe(III)-(oxy)hydroxide, with an initially strong decrease (−68 %) as pH increases, and a subsequent strong positive variation (+59 % with respect to the concentration at CAS1) as pH drops below 5.4 (Fig. 4c). Also, As shows a distribution linked to sorption by Fe(III)-(oxy)hydroxide, but as an oxyanion, it remains adsorbed at an acidic pH (e.g., Frau et al. 2008, 2010), with a negative variation of −96 %. Similar to As, Mo and Sb exhibit decreases of −65 and −82 %, respectively (Fig. 4d).

Chemistry changes in stream water along the Rio Irvi sampled in June 2009
4.3 Characterization of Precipitates
Abundant precipitation of solid phases occurs at the Casargiu outflow and in the Rio Irvi along its course to the sea (Figs. 5a, b, e). XRD analysis of these precipitates showed the presence of amorphous Fe(III)-(oxy)hydroxides, evolving to low-crystalline goethite, occurring as fresh and more consolidated ochreous precipitates (Fig. 6). The color of the wet ochreous precipitates was evaluated in the field using the Munsell Soil Color Charts (1994); all samples were classified as 2.5YR 4/8.

Precipitates in the Casargiu-Rio Irvi system. a Satellite image of a short reach of the Rio Irvi, near the confluence with the Rio Piscinas and about 2 km from the mouth, showing the ochreous precipitates covering the streambed. b Ochreous precipitate at the Casargiu gallery. c The Rio Irvi at the CAS4 sampling point where the high rate of flocculation of green rust (GR) imparts a peculiar green color to stream water, contrasting with the ochreous color of the streambed. d Colloidal dark-green material (GR) retained on a 0.4-μm filter upon water filtration at the CAS4 sampling point. e Ochreous precipitate covering the streambed at CAS9

XRD patterns of precipitates collected at Casargiu and in the Rio Irvi. The fresh ochreous precipitate (upper pattern) was sampled at the exit of the Casargiu gallery (CAS1). The low-crystalline goethite (medium pattern) represents a moderately consolidated, ochreous precipitate collected from the streambed at CAS4. The fresh green precipitate (lower pattern) was made of flocculating colloidal particles retained onto a 0.4-μm filter upon stream water filtration at CAS4; reflections from the basal plane of the sulfate form of green rust are reported
Interestingly, green rust (GR) precipitation has been observed in the field and identified by XRD (Fig. 6); it occurs as flocculating colloidal particles (Fig. 5c) and very thin (2–3 mm) layers just below the surface of the ochreous deposits, especially in the CAS3–CAS5 reach of the Rio Irvi where the high rate of flocculation of this phase imparts a peculiar green color to the stream water, contrasting with the ochreous color of the streambed (Fig. 5c).
Green rusts are mixed ferrous/ferric layered double hydroxides with various anions (e.g., chloride, sulfate, carbonate) and water molecules occupying the interlayer between the iron hydroxide layers (Bearcock et al. 2006; Guilbaud et al. 2013). They typically form under weakly acidic to alkaline conditions in sub-oxic environments and, when exposed to oxic conditions, rapidly transform to various iron (oxy-hydr)oxides. The occurrence of GR in the Rio Irvi has been demonstrated by analyzing the flocculating colloidal material retained on the 0.4-μm filter upon water filtration (Fig. 5d). Although this material showed a color change from dark green to dark orange during the short period of storage (less than 12 h) before analysis, its typical layered structure was preserved as shown by its XRD pattern (Fig. 6), with characteristic peaks (2θ° = 8.12, d 001 = 10.88 Å; 2θ° = 16.36, d 002 = 5.42 Å; 2θ° = 24.53, d 003 = 3.63 Å) representing reflections from the basal plane of the sulfate form of GR (Simon et al. 1997). The ideal formula of sulfate-bearing GR is Fe II4 Fe III2 (OH)12 SO 4 ⋅ 8H 2 O (Simon et al. 2003), but in naturally occurring GRs, the Fe(II)/Fe(III) ratio is known to vary between 0.5 and 3 (typically, it is ∼2 for sulfate GR), and the number of water molecules is also variable (Ahmed et al. 2008). Being an unstable phase under atmospheric conditions, there is no persistent accumulation of GR in the Rio Irvi streambed, even where stream water is clearly oversaturated with respect to GR (see Section 4.4). A transformation into poorly crystalline goethite can be hypothesized, according to the following example reaction for an anhydrous GR (Simon et al. 2003):
where some Fe(II) and all sulfate are released into solution, partly explaining the persistence of ferrous iron in stream water for several kilometers and the near-constant concentration of dissolved sulfate downstream (Fig. 4b). Also, GR potentially acts as a temporary sink for divalent metals (e.g., Zn, Cd, Ni, Co, Mg), which may isomorphically substitute for Fe(II) (Ahmed et al. 2008), because it undergoes incongruent dissolution/oxidative transformation, with subsequent release of metals to water and/or their possible partial adsorption by the newly formed Fe(III)-(oxy)hydroxide. Moreover, it is well known that aqueous Fe(II) induces transformation/recrystallization of Fe(III)-(oxy)hydroxides (Pedersen et al. 2005) and, thus, both GRs and Fe(III)-(oxy)hydroxides have to be considered as dynamic phases that may change composition and structure when exposed to variable pH-Eh conditions.
Table 4 reports the chemical composition of ochreous precipitates collected at the same sampling sites of waters. All solid samples represent fresh precipitates, with the exception of sample CAS2s that is a semi-consolidated precipitate. Most of samples were not pure, often containing variable amounts of detrital minerals (e.g., quartz, illite). Other than Fe, the most abundant elements are S and Zn, followed by Pb, Al, As, and Mn. Each element shows some local concentrations that deviate more or less from the general trend; however, it is possible to recognize an increasing trend for S from CAS1s to CAS9s, and decreasing trends for Zn, Cd, and As, while Pb exhibits no clear trend. Similar results are obtained when concentrations are normalized to Fe (data not shown). To estimate the preferential affinity of an element for the aqueous phase with respect to the solid phase and vice versa, a sort of partition coefficient, similar to an enrichment factor (EF), was calculated as follows:
where El is a generic element and Fe is total iron concentration.
Table 4 reports the average EF values; EF > 1 indicates affinity for the aqueous phase (S, Zn, Cd, Co, Mn) and thus a possible low to moderate removal from water, while EF < 1 is symptomatic of a preferential affinity for the solid phase (Pb, Al, As) and thus a possible moderate to high removal from water, according to data discussed in Section 4.2. Figure 7 shows the variation of EF with distance for two main “hydrophilic” elements (S as sulfate and Zn, Fig. 7a) and two main “hydrophobic” elements (Pb and As, Fig. 7b) in the geochemical conditions occurring at the Casargiu-Rio Irvi system. In particular, EFS exhibits a slightly negative trend, while EFZn shows a moderate positive trend, in accordance with the opposite behavior of anions and cations in sorption processes with Fe(III)-(oxy)hydroxide at observed stream pH values. Similarly, EFAs and EFPb exhibit negative and positive trends, respectively. In fact, As as the As(V)-protonated oxyanion (see Section 4.4) is accumulated in the Fe(III)-(oxy)hydroxide over the entire pH range of sampled waters (average EFAs = 0.12), but the variation of its EF with distance indicates a greater affinity for the solid phase as pH decreases to more acidic values. On the contrary, although Pb also is accumulated in the ochreous precipitates (average EFPb = 0.19), the variation of its EF with distance shows a clear increase towards values close to 1 at acidic pH (EFPb = 0.76 at the last sampling point), which indicates a decreased affinity for the solid phase when partial desorption of Pb from Fe(III)-(oxy)hydroxide occurs at pH < 5.

Variation of enrichment factor (EF) with distance compared to pH variation. a Elements S and Zn preferring the liquid phase (EF > 1). b Elements Pb and As preferring the solid phase (EF < 1)
4.4 Speciation-Solubility Calculations
Aqueous speciation calculated with PHREEQC confirmed dissolved Fe to be dominated in all water samples by Fe(II) species (1.6–3.3 · 10−3 molality), which are 2–3 orders of magnitude higher than Fe(III) species (6.0 · 10−6 to 4.3 · 10−5 molality). The main aqueous species of Zn are Zn2+ (51–53 %) and ZnSO4° (38–41 %), while the speciation of dissolved Pb is more diverse (27–35 % Pb2+, 47–58 % PbSO4°, 5–6 % Pb(SO4)2 2−, 0–11 % PbHCO3 +, 0–10 % PbCO3°). Arsenic is mainly present as H2AsO4 − (74–99 %).
For solubility calculations, the thermodynamic database wateq4f.dat included in PHREEQC was augmented with literature data for the solubility products (K sp) of ochreous Fe(III) phases and sulfate-bearing green rust. The following equilibrium reactions for two-line ferrihydrite and schwertmannite from Majzlan et al. (2004) were added to the database:
where log K sp = 3.7 is the mean value for the range of 3.4–4.0 proposed by Majzlan et al. (2004). It is well known that thermodynamic data for these poorly crystalline phases show a large spread of values (Majzlan et al. 2004 and references therein), and also their stability fields in various pH-pe diagrams are sometimes discordant. As an example, according to Majzlan et al. (2004), schwertmannite is thermodynamically favored over ferrihydrite over a wide range of pH (2–8) when the system contains even small concentrations of sulfate. However, field observations often contradict these statements, as is the case of this study in which schwertmannite was never found in the precipitates of the Casargiu-Rio Irvi system despite the high sulfate concentrations.
The thermodynamic data of GRs are rather uncertain. In particular, the solubility product of sulfate GR cannot be determined directly, according to the following reaction:
(y is the number of interlayer water molecules) because of the extremely low solubility of Fe3+. Therefore, it is necessary to combine reaction (9) with another reaction in which the activity of Fe3+ is controlled by the solubility of a formed ferric phase such as ferrihydrite, introducing further uncertainty due to the variable solubility of poorly crystalline phases as a function of several factors (chemical composition, crystallinity, purity, particle size; Ayala-Luis et al. 2008 and references therein). Also, the difficult determination of the precise number of water molecules per formula unit of GR has represented a problem in estimating reliable thermodynamic data. However, Ayala-Luis et al. (2008) used a novel method to determine the standard Gibbs energy of formation of crystalline sulfate GR based on a slow acid titration with the formation of crystalline magnetite. This method produced an estimate of log K sp of −139.2 ± 4.8 at 25 °C that is invariable with y in reaction (9). Taking into account the low crystallinity of the sulfate-bearing GR collected in the Rio Irvi (Fig. 6), the upper value (log K sp = −134.4) was added to the thermodynamic database wateq4f.dat for reaction (9) with y = 8.
Figure 8 shows the variation of saturation index (SI) with distance for some important minerals in the Casargiu-Rio Irvi system. A general undersaturation occurs for sulfate minerals such as anglesite, bianchite (Fig. 8a), goslarite, melanterite, and epsomite (latter three not shown), while waters are close to equilibrium with respect to gypsum (Fig. 8a). Among the ochreous Fe(III) phases, a generic amorphous ferric hydroxide (already included in the wateq4f.dat database with log K sp = 4.89) is the most compatible with the mineralogical and geochemical data collected in this study; a moderate oversaturation (SI = 1.5–2.3) initially occurs in the reach of the Rio Irvi where pH is circumneutral (CAS1–CAS4), with a gradual trend towards equilibrium where pH becomes acidic (Fig. 8a). Schwertmannite and two-line ferrihydrite exhibit similar SI trends, but with a general oversaturation.

Variation of saturation index with distance for some important minerals in the Casargiu-Rio Irvi system. a Sch, chwertmannite; Fhy, two-line ferrihydrite; Afh, amorphous ferric hydroxide; Gyp, gypsum; Ang, anglesite; Bia, bianchite. b Sid, siderite; Smi, smithsonite; Cer, cerussite; Cal, calcite. c Gr, sulfate-bearing green rust
As hypothesized in Section 4.2 with reaction (2), the outflow from the Casargiu gallery (CAS1) is close to equilibrium with siderite (SI = 0.3), and smithsonite (SI = −0.1) as well (Fig. 8b); equilibrium is maintained until CAS4, and then the pH decrease results in steep undersaturation. Waters are clearly undersaturated with respect to calcite and cerussite (Fig. 8b).
In agreement with field observations, Fig. 8c shows that there is no precipitation of sulfate GR at CAS1 (SI = −1.4); then a moderate oversaturation occurs at CAS2–CAS3 (SI = 1.0–0.9) as soon as water comes in contact with the air and ferrous iron begins to oxidize to ferric iron (see Section 4.2). As a matter of fact, a first flocculation of GR has been observed in the reach between CAS2 and CAS3, but GR precipitation becomes evident only at CAS3 and reaches its maximum at CAS4, as indicated by the SI value of 4.8. Flocculation of GR continues up to CAS5 (SI = 1.0), and then a strong undersaturation occurs from CAS6 (SI = −2.3) to CAS9 (SI = −14.2) as pH drops below 5.6 (Fig. 8c).
4.5 Discharge of Metals to the Mediterranean Sea
The estimated amount of dissolved metals discharged daily from the Rio Irvi into the Mediterranean Sea is reported in Table 5. These calculations derive from dissolved concentrations measured in the stream sampled about 1 km upstream of its mouth (CAS9). Assuming the lowest flow measured in the stream in 2008 (20 L/s) as the mean annual flow, the amount of metals discharged in the year 2008 into the Mediterranean Sea was estimated at 328, 1.8, 0.5, and 0.3 ton of Zn, Ni, Cd, and Pb. Compared to the global flux estimation of dissolved metals from uncontaminated rivers to the oceans (Gaillardet et al. 2003; Zn 23,000 ton/year, Cd 3000 ton/year, Pb 3000 ton/year), the contribution of Zn from the Casargiu mine drainage to the sea would correspond to 1.4 % of the global riverine annual flux of dissolved Zn. A similar percentage (1.6 %) is obtained when considering riverine dissolved Zn flux to the oceans (20,000 ton/year) by Poulton and Raiswell (2000). This is a conservative estimation because metal loads increase significantly with flow, as shown in Table 5 for April and June 2009, leading to Zn discharges to the Mediterranean Sea that would correspond to 4–6 % of the global riverine annual flux of dissolved Zn according to both data sources. For Cd and Pb, the percentages are always below 0.1 %, regardless of whether low or high flow estimates are used in the calculation.
The flux of dissolved Zn from uncontaminated rivers to the Western Mediterranean Sea was estimated to be 130 ton/year by Elbaz-Poulichet et al. (2001) considering only the Rhone (1,710,000 L/s) and Ebro (626,000 L/s) rivers that represent 70 % of the total river discharge (in terms of water volume) to the Western Mediterranean Sea. According to these partial data, the estimated discharge of dissolved Zn from the Rio Irvi (328 ton/year in 2008 assuming a constant flow of 20 L/s) to the sea would be 2.5 times higher than the total amount delivered from the Rhone and Ebro Rivers to the Western Mediterranean Sea.
5 Conclusions
The geochemical-mineralogical approach taken in this study appeared to be a valuable tool for the understanding of contamination processes occurring at an abandoned mining site. This study shows that drainages from flooded mines are significant mining-related sources of contamination. In particular, near-neutral drainages may be highly contaminated and represent a major risk to the hydrographic system, heavily modifying water chemistry and composition of stream sediments.
The most important results of this study can be summarized as follows: (1) the impact on the Rio Irvi of the near-neutral drainage from the flooded Casargiu mine (Sardinia, Italy) was evaluated to be very high since the first discharge in 1997; (2) the temporal variations in chemistry of the mine water discharge were studied in the period 1997–2012 and showed that the period of flushing is still too brief to observe a substantial lowering of the pollution level; (3) the chemistry changes along the Rio Irvi, from the discharge point (the Casargiu gallery) downstream over a distance of 6 km, were fully studied for the first time in 2009, and their modeling showed various geochemical-mineralogical processes taking place either simultaneously or sequentially; and (4) a high discharge of dissolved metals from the Rio Irvi to the Mediterranean Sea was estimated.
In more detail, the water flowing out of the Casargiu gallery is circumneutral (pH in the range of 5.8 to 6.2) but has very high concentrations of sulfate and divalent metals (especially Zn, Fe(II), Mn, Cd, Co, Ni, Pb). Even though concentrations of most metals have decreased somewhat compared with values recorded at the first stages of rebound and discharge at Casargiu, the concentrations still exceed toxic threshold levels at this site after more than 15 years of flushing.
Mine drainage water from Casargiu (20–70 L/s) flows into the Rio Irvi, which is uncontaminated upstream of the Casargiu discharge. A significant decrease in dissolved Fe occurs downstream by precipitation of solid phases (amorphous and poorly crystalline Fe(III)-(oxy)hydroxide, and sulfate-bearing green rust). The precipitation of green rust from stream water is an uncommon and interesting phenomenon, described and modeled in this study. The gradual decrease of pH downstream is accompanied by large removal of Pb and As from water by adsorption; however, Pb is desorbed as pH drops below 5. In contrast, Zn and Cd concentrations in the Rio Irvi decrease only 6 and 15 % compared to the Casargiu source. As a consequence, even very small streams are able to discharge high metal loads to the sea, as is the case of the Rio Irvi flowing into the Mediterranean Sea.
After the failure of the treatment plant built in 2007 (Concas et al. 2006), this study suggests that construction of a new treatment plant that takes into account the geochemical-mineralogical processes described in this study could help remediate the extremely contaminated drainage at Casargiu before discharge into the Rio Irvi. The solid residues produced by the treatment plant should be properly disposed. Monitoring of the treated effluent also would verify the efficiency of the future treatment plant.
References
Adams, R., & Younger, P. L. (2001). A strategy for modeling ground water rebound in abandoned deep mine systems. Ground Water, 39(2), 249–261.
Ahmed, I. A. M., Shaw, S., & Benning, L. G. (2008). Formation of hydroxysulphate and hydroxycarbonate green rusts in the presence of zinc using time-resolved in situ small and wide angle X-ray scattering. Mineralogical Magazine, 72(1), 159–162.
Ayala-Luis, K. B., Koch, C. B., & Hansen, H. C. B. (2008). The standard Gibbs energy of formation of Fe(II)Fe(III) hydroxide sulfate green rust. Clays and Clay Minerals, 56(6), 633–644.
Bain, J. G., Blowes, D. W., Robertson, W. D., & Frind, E. O. (2000). Modelling of sulfide oxidation with reactive transport at a mine drainage site. Journal of Contaminant Hydrology, 41, 23–47.
Bain, J. G., Mayer, K. U., Blowes, D. W., Frind, E. O., Molson, J. W. H., Kahnt, R., & Jenk, U. (2001). Modelling the closure-related geochemical evolution of groundwater at a former uranium mine. Journal of Contaminant Hydrology, 52, 109–135.
Banks, D., Younger, P. L., Arnesen, R.-T., Iversen, E. R., & Banks, S. D. (1997). Mine-water chemistry: the good, the bad and the ugly. Environmental Geology, 32(3), 157–174.
Bearcock, J. M., Perkins, W. T., Dinelli, E., & Wade, S. C. (2006). Fe(II)/Fe(III) ‘green rust’ developed within ochreous coal mine drainage sediment in South Wales, UK. Mineralogical Magazine, 70(6), 731–741.
Biddau, M. (1978). Indagine chimica-idrologica sulle possibilità di utilizzazione delle acque edotte nel Sulcis-Iglesiente. La Programmazione in Sardegna, 67(68), 99–115 (in Italian).
Biddau, R., Da Pelo, S., & Dadea, C. (2001). The abandoned mining area of Montevecchio-Ingurtosu. Rendiconti Seminario Facoltà Scienze Università Cagliari, 71(2), 109–123.
Caboi, R., Cidu, R., Fanfani, L., Lattanzi, P., & Zuddas, P. (1999). Environmental mineralogy and geochemistry of the abandoned Pb-Zn Montevecchio-Ingurtosu mining district, Sardinia, Italy. Chronique Recherche Minière, 534, 21–28.
Cidu, R., & Fanfani, L. (2002). Overview of the environmental geochemistry of mining districts in southwestern Sardinia, Italy. Geochemistry: Exploration, Environment, Analysis, 2, 243–251.
Cidu, R., Biddau, R., & Nieddu, G. (2007). Rebound at Pb-Zn mines hosted in carbonate aquifers: influence on the chemistry of groundwater. Mine Water and the Environment, 26(2), 88–101.
Concas, A., Ardau, C., Cristini, A., Zuddas, P., & Cao, G. (2006). Mobility of heavy metals from tailings to stream waters in a mining activity contaminated site. Chemosphere, 63, 244–253.
Cravotta, C. A., III. (2007). Passive aerobic treatment of net-alkaline, iron-laden drainage from a flooded underground anthracite mine, Pennsylvania, USA. Mine Water and the Environment, 26, 128–149.
Elbaz-Poulichet, F., Guieu, C., & Morley, N. M. (2001). A reassessment of trace metal budgets in the Western Mediterranean Sea. Marine Pollution Bulletin, 42(8), 623–627.
Frau, F. (2000). The formation-dissolution-precipitation cycle of melanterite at the abandoned pyrite mine of Genna Luas in Sardinia, Italy: environmental implications. Mineralogical Magazine, 64(6), 995–1006.
Frau, F., Biddau, R., & Fanfani, L. (2008). Effect of major anions on arsenate desorption from ferrihydrite-bearing natural samples. Applied Geochemistry, 23(6), 1451–1466.
Frau, F., Addari, D., Atzei, D., Biddau, R., Cidu, R., & Rossi, A. (2010). Influence of major anions on As(V) adsorption by synthetic 2-line ferrihydrite. Kinetic investigation and XPS study of the competitive effect of bicarbonate. Water, Air, and Soil Pollution, 205, 25–41.
Gaillardet, J., Viers, J., & Dupré, B. (2003). Trace elements in river waters. In J. I. Drever (Ed.), Surface and Ground Water, Weathering and Soils, vol. 5 (pp. 225–272). In H. D. Holland & K. K. Turekian (Eds.), Treatise on Geochemistry, Elsevier–Pergamon, Oxford.
Gandy, C. J., & Younger, P. L. (2007). Predicting groundwater rebound in the South Yorkshire Coalfield, UK. Mine Water and the Environment, 26(2), 70–78.
Geldenhuis, S., & Bell, F. G. (1997). Acid mine drainage at a coal mine in the eastern Transvaal, South Africa. Environmental Geology, 34(2/3), 234–242.
Gray, N. F. (1997). Environmental impact and remediation of acid mine drainage: a management problem. Environmental Geology, 30(1/2), 62–71.
Guilbaud, R., White, M. L., & Poulton, S. W. (2013). Surface charge and growth of sulphate and carbonate green rust in aqueous media. Geochimica et Cosmochimica Acta, 108, 141–153.
Gzyl, G., & Banks, D. (2007). Verification of the “first flush” phenomenon in mine water from coal mines in the Upper Silesian Coal Basin, Poland. Journal of Contaminant Hydrology, 92, 66–86.
Iribar, V. (2004). Origin of neutral mine water in flooded underground mines: an appraisal using geochemical and hydrogeological methodologies. In Mine Water 2004 – Process, Policy, and Progress, Proceedings of International Mine Water Association Symposium 2004, Newcastle upon Tyne, United Kingdom 20–25 September 2004, ISBN: 978-1-62276-985-8 (pp. 260–274).
Majzlan, J., Navrotsky, A., & Schwertmann, U. (2004). Thermodynamics of iron oxides: part III. Enthalpies of formation and stability of ferrihydrite (∼Fe(OH)3), schwertmannite (∼FeO(OH)3/4(SO4)1/8), and ε-Fe2O3. Geochimica et Cosmochimica Acta, 68(5), 1049–1059.
Munsell Soil Color Charts (1994). Revised edition. Gretag Macbeth, New Windsor, NY.
Palache, C., Berman, H., & Frondel, C. (1944). Dana’s system of mineralogy (7th ed., Vol. I). New York: John Wiley and Sons.
Palache, C., Berman, H., & Frondel, C. (1951). Dana’s system of mineralogy (7th ed., Vol. II). New York: John Wiley and Sons.
Parkhurst, D. L., & Appelo, C. A. J. (1999). User’s guide to PHREEQC (version 2)—a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. USGS Water-Resources Investigations Report 99–4259, Denver, CO, USA.
Pedersen, H. D., Postma, D., Jakobsen, R., & Larsen, O. (2005). Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II). Geochimica et Cosmochimica Acta, 69(16), 3967–3977.
Poulton, S. W., & Raiswell, R. (2000). Solid phase associations, oceanic fluxes and the anthropogenic perturbation of transition metals in world river particulates. Marine Chemistry, 72(1), 17–31.
Ptacek, C. J., & Blowes, D. W. (1994). Influence of siderite on the pore-water chemistry of inactive mine-tailings impoundments. In C. N. Alpers & D. W. Blowes (Eds.), Environmental geochemistry of sulphide oxidation, ACS symposium series 550, Chapter 13 (pp. 172-189).
RAS (2003). Piano regionale di gestione dei rifiuti. Regione Autonoma della Sardegna, 255 pp. (in Italian).
RAS (2013). Serie storica delle altezze di precipitazione giornaliera dal 1922 al 2010. Available at: http://www.regione.sardegna.it/j/v/25?s=131338&v=2&c=5650&t=1. Accessed 5 January 2015.
Razowska, L. (2001). Changes of groundwater chemistry caused by the flooding of iron mines (Czestochowa Region, Southern Poland). Journal of Hydrology, 244, 17–32.
Rosner, U. (1998). Effects of historical mining activities on surface water and groundwater—an example from northwest Arizona. Environmental Geology, 33(4), 224–229.
Royle, M. (2007). Giant mine: identification of inflow water types and preliminary geochemical monitoring during reflooding. Mine Water and the Environment, 26(2), 102–109.
Simon, L., Gènin, J.-M. R., & Refait, P. (1997). Standard free enthalpy of formation of Fe(II)-Fe(III) hydroxysulphite green rust one and its oxidation into hydroxysulphate green rust two. Corrosion Science, 39(9), 1673–1685.
Simon, L., François, M., Refait, P., Renaudin, G., Lelaurain, M., & Génin, J.-M. R. (2003). Structure of the Fe(II-III) layered double hydroxysulphate green rust two from Rietveld analysis. Solid State Sciences, 5, 327–334.
WHO. (2006). Guidelines for drinking-water quality (3rd ed.). Geneva: World Health Organization.
Wood, S. C., Younger, P. L., & Robins, N. S. (1999). Long-term changes in the quality of polluted minewater discharges from abandoned underground coal workings in Scotland. Quarterly Journal of Engineering Geology and Hydrogeology, 32, 69–79.
Younger, P. L. (2000). Predicting temporal changes in total iron concentrations in groundwaters flowing from abandoned deep mines: a first approximation. Journal of Contaminant Hydrology, 44, 47–69.
Younger, P. L. (2001). Mine water pollution in Scotland: nature, extent and preventative strategies. Science of the Total Environment, 265, 309–326.
Acknowledgments
This study was financially supported by RAS (Sardinian Regional Government), Fondazione Banco di Sardegna, University of Cagliari (ex-60 % funds to F. Frau and R. Cidu), and MIUR (Italian Minister of University and Research) (PRIN-2009 funds to R. Cidu; PRIN-2010 funds to P. Lattanzi). The Mineral Resources Program of the US Geological Survey provided funds for R.B. Wanty. The authors wish to thank G. Contis for IC analyses. The use of brand names in this paper is for descriptive purposes only and does not constitute endorsement by the US Government or Italian academic institutions. Data used to produce the results of this paper is archived at the Department of Chemical and Geological Sciences, University of Cagliari, Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Frau, F., Medas, D., Da Pelo, S. et al. Environmental Effects on the Aquatic System and Metal Discharge to the Mediterranean Sea from a Near-Neutral Zinc-Ferrous Sulfate Mine Drainage. Water Air Soil Pollut 226, 55 (2015). https://doi.org/10.1007/s11270-015-2339-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-015-2339-0