
Abstract
The complete mineralization of the conazole fungicide triadimefon in water at pH 3 using electrochemical advanced oxidation processes, electro-Fenton and photo electro-Fenton, was achieved. The electrochemical system consisted of a one-compartment electrochemical cell of 100 mL provided with a glassy carbon mesh electrode (cathode) and a concentric outer steel mesh as anode. The electrolysis was realized at constant current. The most remarkable features are as follows: (1) photo electro-Fenton process reaches a complete mineralization of triadimefon after 2 h of electrolysis with respect to electro-Fenton method; and (2) 4-chlorophenol, hydroquinone, carboxylic acids, and inorganic ions were detected as intermediates of degradation processes, which end with the complete mineralization of triadimefon to CO2 + H2O. (3) A reaction pathway for the oxidation of triadimefon fungicide by hydroxyl radicals that accounts for almost all detected intermediates is proposed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Triadimefon (Table 1) is a compound widely used as agrochemical systemic fungicide due to its large spectrum of application and be very cheap. This compound belongs to the family of conazoles, a large family of synthetic triazole fungicides used extensively in agriculture to prevent fungal growth on fruits, vegetables, cereals, and seeds. In Chile, triadimefon is used to prevent, cure, or eradicate fungal mainly in vineyards and apple orchards. The formulation consists of 25 % triadimefon (25 g/kg) and its application is extended from 7 to 45 days depending on the crop and the degree of progress of the attack of the fungus. It is also used in pharmacology to treat local and systemic fungal infections, or to prevent serious fungal infections in cancer patients with compromised immune systems (Chen et al. 2009).
Triadimefon is a moderately toxic compound that carries the signal word WARNING on its label, has an acute oral LD50 in rats of 569 mg/kg, around 1,000 mg/kg in mice, and about 500 mg/kg in rabbits and dogs (Kidd and James 1991). Exposure of rodents to high concentrations of conazole fungicides has resulted in multiple toxic endpoints, including carcinogenic, neurological, reproductive, and endocrine effects (Allen et al. 2006). So it is not uncommon to find triadimefon dissolved in waters feasible to be used for irrigation or drinking water sources. Hence, the interest in conducting a study of degradation of this compound using new and effective experimental methods.
The electrochemical technologies appear like an effective alternative in wastewater remediation due to the new procedures that have been implemented in the later years (Poyatos et al. 2010), more of which have been used now in industrial process such as electrocatalysis. The main electrochemical reagents, the electrons, are a clean reagent allowing environmental compatibility and moreover versatility, high energy efficiency, amenability of automation, and safety because it operates at mild conditions. The electrochemical advanced oxidation processes (EAOPs) (Brillas et al. 2009; Martínez-Huitle and Brillas 2009; Sirés et al. 2007) such as electro-Fenton (EF) and photo electro-Fenton (PEF) belong to a group of new electrochemical alternatives technologies for the remediation of organic pollutants in wastewaters. These methods have in common the generation of ●OH radicals from hydrogen peroxide generated via reduction of oxygen (Eq. (1)) through the classical Fenton’s reaction (Quiang et al. 2002). The ●OH radicals are one of the best species capable for destroying almost all kinds of organic contaminants (Cañizares et al. 2006). In EF process, the generation of ●OH occurs from the reaction between H2O2 activated in acidic effluents with Fe2+ ion as catalyst (Fenton’s reagent) according to reaction (2):
When using the EF method, the amount of Fe2+ added is much smaller than in traditional Fenton method because in the electrochemical process, Fe2+ is regenerated at the cathode.
If electrolyte solution is irradiated with UV light (PEF process), the generation of homogeneous ●OH is favored together with the photodegradation of complexes of Fe(III) with organic compounds (Salazar et al. 2011; Ting et al. 2008) as follows:
Regarding the electrochemistry of triadimefon, conazole compounds, or other similar fungicides, within what we are aware of, the literature is very poor. Recently the use of triadimefon was reported as inhibitor of copper corrosion in seawater at different electrolytes (Hu et al. 2010). In another work, the authors developed a very sensitive electroanalytical method for the determination of ketoconazole, an imidazole derivative, using differential pulse voltammetry at glassy carbon electrode in ammonium buffer (pH 9), reporting a detection limit of 4.0 × 10−11 mol L–1 (Peng et al. 2001). The electrochemistry or remediation of contaminated water with triadimefon was recently reported on the removal of triadimefon in a municipal sewage treatment plant in Greece (Stamatis et al. 2010). The global process used in this work includes three steps: mechanical (primary), activated sludge (secondary), and sand filtration and chlorination (tertiary), as is usually applied in Greece. The authors report 65 % mean removal for triadimefon after secondary treatment and 93 % after tertiary treatment.
Thus, the aim of this work is to determine the laboratory experimental conditions in which one of these methods (EF and PEF) shows the better performance for the electro-mineralization of triadimefon in aqueous solution with the idea that in the near future these results are extrapolated to a pilot plant. In this sense, it is very important in this work to identify intermediates formed during the process—aromatic compounds, carboxylic acids, and inorganic ions—and to develop a kinetic analysis and determine the efficiency of both processes. The advantage of using the EF and PEF methods is that as the ●OH radicals formed are so active, the degradation of the pesticide is almost total in short time.
2 Materials and Methods
2.1 Chemicals and Reagents
Triadimefon (99.7 % PESTANAL®) was purchased from Sigma-Aldrich, and Na2SO4, FeSO4·7H2O, H2SO4, and NaOH (Merck, West Point, PA, USA) were all used as received. The other chemicals such as oxalic, maleic, pyruvic, oxamic, pivalic, acetic, and fumaric acids were also purchased from Sigma-Aldrich or Merck and used as received. O2 (99.99 %, Indura S.A.) was bubbled directly into the solution. These were prepared by dissolving 50 mg of triadimefon in 1 L of 0.1 M Na2SO4 at pH 3 in double distilled water (conductivity <6 μS). The pH, measured using an EXTECH Instrument 321990 pH meter, was adjusted with H2SO4 or NaOH concentrated solutions.
2.2 Electrochemical Processes
The electrolysis were carried out at 35 °C (Boye et al. 2002) under controlled current using a glassy carbon mesh electrode (CH Instrument) as cathode, and a concentric outer steel mesh as anode, each one with an exposed area of 25 cm2, in the presence of pure oxygen bubbled across the solution. The electrolyte was 0.1 M Na2SO4 at pH 3 and variable Fe2+ concentration, in a one-compartment cell of 100 mL using a WENKING POS 75 potentioscan (Resistance 1 Ω), under magnetic stirring.
In addition to the above methods for PEF, the system was irradiated using a medium-pressure Hg lamp without filter with a wavelength of 360 nm.
2.3 Total Organic Carbon (TOC) Determination
In order to know the degree of degradation of triadimefon, total carbon analysis was performed on electrolyte samples obtained during the electrolysis using a Shimadzu 5050 TOC Analyzer.
With the TOC decay data, the mineralization current efficiency (MCE) for each treated solution was estimated by Eq. (5) (Salazar et al. 2011):
where n is the number of electrons consumed in the mineralization of each dye, F is the Faraday constant (96,487 C mol−1), V s is the solution volume (dm3), is Δ(TOC)exp is the experimental TOC decay (mg dm−3), 4.32 × 107 is a conversion factor (3,600 s h−1 × 12,000 C mol−1), m = 16, corresponding to the number of carbon atoms in the triadimefon molecule, I is the applied current (A), and t is the electrolysis time (h). The n value was taken as 58, considering the complete mineralization to CO2 and NH +4 ion from the following reaction:
2.4 High-Performance Liquid Chromatography (HPLC)
The residual triadimefon concentration and detection of its degradation products were performed by HPLC using a Waters 600 chromatograph equipped with a 600 controller pump and a 996 photodiode array (PDA). The acquisition and data processing were done with the Millenium-32 software. A Kromasil C-18 chromatographic column (4.6 × 150 mm2) together with a C-18 Bondapak precolumn (30 × 4.6 mm2) was employed. The injector was a 20-μL Rheodyne valve. Samples of electrolyzed solutions were filtered through 0.45-μm PTFE filters (Whatman) before analysis. A mixture of 80 % methanol and 20 % H2O at 1 mL min−1 was used as carrier (Zhou et al. 2007). The triadimefon decay was determined at the maximum wavelength of its UV absorption band ?λ = 277 nm.
Analytical determination of generated carboxylic acids was followed by ion exclusion chromatography injecting 20-μL samples into the HPLC chromatograph fitted with an Aminex HPX-87 H, 300 × 7.8 mm, column at 35 °C from Bio-Rad and coupled with the photodiode array detector selected at 210 nm, using a 4 mM H2SO4 solution at 0.6 mL min−1.
Released inorganic ions were quantified by ionic chromatography by injection of 50-μL aliquots in a Waters 600 chromatograph equipped with a 600 controller pump and a Waters 431 conductivity detector. The Cl− concentration in electrolyzed solutions was determined using an IC-Pak AHR, 75 mm × 4.6 mm (i.d.), anion column at 35 °C and a mobile phase of borate gluconate at 1.0 mL min−1. The NH +4 concentration was determined with an IC-Pak C M/D, 150 mm × 3.9 mm (i.d.), cation column at 25 °C and a mobile phase of 0.1 mM EDTA/3.0 mM HNO3 at 1.0 mL min−1.
2.5 Gas Chromatography–Mass Spectrometry (GC–MS)
Detection and identification of aromatic intermediate was also performed by three times extraction with 100 mL of CH2Cl2. The collected organic solutions were dried with anhydrous Na2SO4, filtered, and evaporated to about 3 mL using a rotary evaporator for further separation and identification by GC–MS. A gas chromatograph/mass selective detector (5890/5972) combination (Hewlett-Packard, Palo Alto, CA, USA) and a Hewlett-Packard 7673 autosampler were used for the analyses. The m/z range monitored was 45–400 with a scan rate of 1 scan/s; the normal energy electron was set at 70 eV. A Hewlett-Packard Ultra-1 column, 25 cm with 0.2 mm i.d. film thickness (Little Falls, Wilmington, DE, USA), was used.
3 Results and Discussion
3.1 Degradation of Triadimefon by Electro-Fenton and Photo Electro-Fenton
In order to determine the minimum concentration of Fe2+ ions that supports the largest possible catalytic effect, experiments with different [Fe2+] were made. Figure 1 shows the results for the mineralization of 50 mg L−1 of triadimefon solutions. In the absence of ferrous ions, no degradation of pesticide occurs, evidencing that triadimefon does not react at the electrode surface. In the presence of Fe2+ ions, a maximum TOC decay is observed with 0.1 and 0.5 mM Fe2+ when an electric current of 500 mA has been circulating in the system for 2 h. Consequently, considering that the difference in TOC in the mineralization using 0.1 and 0.5 mM of Fe2+ is not significant, the value of 0.1 mM of Fe2+ was elected as the best for further experiments.

Influence of Fe2+ concentration in TOC decay of 50 mg L−1 of triadimefon in 0.1 M Na2SO4, pH 3 at 35 °C with an applied density current of 20 mA cm−2: filled squares—0.1 mM, filled circles—0.25 mM, filled up-pointing triangles—0.5 mM, filled down-pointing triangles—1.0 mM of Fe2+. Electrodes area = 25 cm2
As was expected, the rate of triadimefon degradation is dependent on the current intensity applied during electrolysis, as is shown in Fig. 2, where the TOC values are plotted as the time electrolysis function for applied current density from 4 mA cm−2 (curve a) to 28 mA cm−2 (curve d). The decay of the total organic carbon (TOC) increases with the increase in applied current density until 28 mA cm−2 (curve c). Higher density currents values (i.e., 28 mA cm−2, curve d) did not increase the organic compound degradation rate, which is evidenced because the dTOC/dt slope in Fig. 2 is independent of the applied density current for I > 20 mA cm−2, which could be attributed to the oxidation process that has reached its maximum possible rate. In other words, there is a diffusion control at the electrode/electrolyte interface, where the ●OH radicals are generated. It could be that the ●OH radicals’ generation rate was the reaction determining step (rds), but this is unlikely because reaction 1 is fast, so it is more likely a diffusional control. Another effect could be a decrease of H2O2 production by its accumulation at the cathode/solution interface and its partial decomposition. Moreover, high currents may favor the hydrogen gas evolution competing for electrons in the formation of H2O2 (Quiang et al. 2002). As can be seen in Fig. 2, TOC decay reaches 55 % at 20 mA cm−2 and 57 % at 28 mA cm−2. For this reason, the other factors that can affect the triadimefon degradation in the electrolysis were determined at a current density of 20 mA cm−2.

Effect of the applied current and UVA radiation (?λ = 360 nm) in TOC decay of 50 mg L−1 of triadimefon in 0.1 M Na2SO4 and 0.1 mM Fe2+at pH 3.0 and 35 °C by EF: filled up-pointing triangles—4 mA cm−2, filled down-pointing triangles—10 mA cm−2, filled circles—20 mA cm−2, filled squares—28 mA cm−2, open circles—PEF process at 20 mA cm−2. Electrodes area = 25 cm2
A large increase in the mineralization rate of triadimefon was obtained when the electrochemical system was irradiated with UVA light (curve d in Fig. 2). Now, the TOC decay is 95 % after 2 h, in accordance with a homogeneous process of generation of ●OH and an increase in the photodegradation of complexes formed by Fe(III) with organics (Kusic et al 2006; Skoumal et al. 2008).
The MCE values (calculated from Eq. (5)) for the comparative degradation of triadimefon by EF and PEF shown in Fig. 2 are depicted in Fig. 3. As expected, higher MCE is obtained using PEF than EF. In EF, a maximum efficiency of 26 % was determined at 20 min and decay to 18 % at the end of the electrolysis. A better maximum for MCE of about 46 % can be observed in Fig. 3 at 20–30 min for the PEF degradation of triadimefon, followed by a fast decrease in efficiency up to ca. 27 % in agreement with TOC decay trend showed in Fig. 2. This behavior in MCE by both methods can be related to the increasing formation and mineralization of organic intermediates that react rapidly with •OH, while at long electrolysis time, the gradual formation of stable by-products, such as carboxylic acids, contribute to the loss of the efficiency in EF. While in PEF, the tendency agrees with the quick TOC removal of about 50 % at the maximum efficiency as a result of the enhancement of organics mineralization by the production of more •OH from reaction (3) and photolysis of Fe(III) complexes by UV radiation.

Variation of mineralization current efficiency calculated from Eq. (6) with electrolysis time for experiments of Fig. 2. Processes: EF (filled circles) and PEF (open circles) for 50 mg L−1 of triadimefon solutions at pH 3.0 and 35 °C at 20 mA cm−2. Wave longitude in PEF = 360 nm
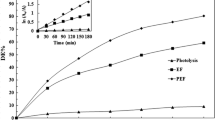
The determination of the triadimefon remaining in the electrolyte solution was made by chromatographic assays, where triadimefon has a retention time (r t ) of 5.1 min under the experimental conditions described in the experimental section of this work. The analysis was performed by sampling the electrolysis cell originally with 50 mg L−1 of triadimefon electrolyzing at 20 mA cm−2, in the absence and the presence of UV irradiation. A decrease in the peak height at r t = 5.1 min was observed. A correlation of the triadimefon concentration (mg L−1) as a function of the electrolysis time is shown in Fig. 4 for both sets of experimental data. The results clearly show that the concentration of triadimefon decreases more quickly by PEF process, reaching a value close to zero after 120 min whereas the fungicide concentration decay was only 72 % in the same electrolysis time when the system has not been irradiated. When the system is irradiated, the mineralization of triadimefon is almost total.

Concentration decay of 50 mg L−1 of triadimefon in 0.1 M Na2SO4 and 0.1 mM Fe2+ at pH 3.0 and 35 °C by EF (filled circles) and PEF (open circles) processes. Electrodes area = 25 cm2. Effect of UVA radiation (?λ = 360 nm) at 20 mA cm−2 of applied current. Insert shows the corresponding kinetic analysis assuming that triadimefon follows a pseudo-first-order reaction
The triadimefon concentration decay is faster than its mineralization, measured by TOC analysis. If we compared Figs. 2 and 3 by HPLC experiments, the specific decay of the pesticide can be seen while by TOC measurements the transformation from organic compound to CO2 is observed. So, for the first time, triadimefon is transformed or degraded to more oxidized organic compounds before its complete mineralization.
The reaction of triadimefon with ●OH radical can be written as
The corresponding kinetic equation is:
where c ●OH and c T are the ●OH and triadimefon concentration, respectively, and k is the intrinsic second-order rate constant. As was commented before, the reaction by which ●OH radicals are formed (reaction 2) is probably faster than reaction (7), and together with the quick regeneration of hydroxyl radical by means of Eq. (3) implies that the ●OH concentration can be considered as constant. Thus, Eq. (8) becomes to a pseudo-first-order kinetics law, written as:
where k app is the experimentally determined rate constant (Chang 2000). The integration of Eq. (9) gives the typical first-order kinetic equation, which allows the evaluation of the rate constant from the slope of a plot of Ln (c 0/c t) against the electrolysis time (insert in Fig. 4). The k app values increase from 1.75 × 10−4 s−1 obtained by EF method to 4.35 × 10−4 s−1 by PEF process increasing 2.5 times with respect to the process without radiation, in concordance with a high efficiency in the ●OH generation process.
3.2 Proposed Degradation’s Sequence in the Mineralization of Triadimefon by PEF
As can be mentioned before, the complete degradation of triadimefon to CO2 occurs with the formation of different products and by products. For this reason, intermediates and/or products formed during the electrolysis were followed by chromatographic techniques in order to identify and quantify these oxidation products, and provide the evidences that allow to propose a possible reaction pathway. Samples obtained at different electrolysis times were extracted with CH2Cl2, concentrated and injected in a GC–MS equipment. The MS spectra displayed the corresponding peaks associated at triadimefon (m/z = 294, M + and 296, M + 2) at t r = 14.4 min, together with two additional peaks associated with the presence of 4-chlorophenol (m/z = 128, M + and 130, M + 2) at t r = 12.6 min and for tetramethylsilane derivative hydroquinone (m/z = 106, M+) at t r = 13.7 min, which correspond to the main aromatic intermediates. This result explains the fact that the TOC decreases are slower than the decreases of the triadimefon concentration.
On the other hand, ion-exclusion chromatograms of the above electrolyzed solutions of triadimefon (20 mA cm−2 by PEF process) exhibited peaks associated with the presence of carboxylic acids formed during the oxidation. To identify which carboxylic acids are present in the chromatograms, samples of pure and mixtures of carboxylic acid were used as pattern, under the same experimental conditions. The results agree with the presence of oxalic (t r = 6.6 min), maleic (t r = 7.9 min), pyruvic (t r = 9.0 min), oxamic (t r = 9.4 min), pivalic (t r = 13.2 min), acetic (t r = 15.4 min), and fumaric (t r = 16.0 min) acids, and its evolution during the electrolysis is shown in Fig. 5a and b. Maleic and fumaric acids come from the oxidation of the aril moiety of aromatics; pivalic and pyruvic acid could be formed by oxidation of 4-chlorophenol, whereas oxamic acid could be formed when the triazol ring is open. At the end of the electrolysis, oxalic and oxamic acids are the later carboxylic acids that are directly transformed into CO2 (Brillas et al. 2009). Under the PEF conditions tested, the Fe(III) complexes of detected acids are expected to be the predominant species in electrolyzed solutions (Ruiz et al. 2011).

Evolution of carboxylic acid detected during the PEF degradation of 50 mg L−1 of triadimefon in 0.1 M Na2SO4 and 0.1 mM Fe2+ under the same conditions of Fig. 3. In (a) are shown pivalic (open triangles), maleic (filled up-pointing triangles), and fumaric (filled down-pointing triangles) acids, and in (b) acetic (open squares), pyruvic (filled squares), oxamic (open circles), and (oxalic) fumaric acids
The loss of NH +4 and NO −3 ions in the mineralization of N-derivatives is very common (Martínez-Huitle and Brillas 2009). For this reason, for triadimefon the presence of both ions by ionic chromatography was verified in the final electrolyzed solutions using PEF. While the presence of NO −3 ion was not detected in such solutions, the presence of 0.46 mmol L−1 (91.0 % of initial N) for the NH +4 ion was found. Since the corresponding remaining oxamic acid only contains 2.2 % of initial N (see Fig. 3), one can infer that a part of the initial N of triadimefon (ca. 6.8 %) is lost as volatile N-species, probably N2 and NOx. The presence of 0.16 mmol L−1 (93.3 % of initial Cl) Cl− ion was found in the finale triadimefon solution.
Thus, through EF and PEF at pH 3, the mineralization of solutions containing triadimefon for the attack of ●OH could begin simultaneously on the bonds N–C–O involving tree possibilities to break the starting molecule, giving for one site 4-cholophenol (way 1 in the proposed pathway, Scheme 1), pivalic acid (2 in the scheme), and oxamic acid by the fast rupture of triazole ring. The 4-chlorophenol molecule could be attacked by a second ●OH giving p-hydroquinone and releasing Cl− ions. p-Hydroquinone reacts again with ●OH, giving a mixture of maleic and fumaric acids, which are subsequently transformed into oxalic acid and finally CO2. By way 2, the attack of ●OH radicals on triadimefon gives, in a first step, pivalic acid. This is attacked by ●OH to give pyruvic acid, acetic acid, and then subsequently oxalic acid. A third route of reaction is also present for triadimefon which would involve the quick formation of oxamic acid. From its oxidation would occur the formation of NH +4 ions and oxalic acid. Finally, oxalic acid is converted into CO2; being the initial molecule, triadimefon fungicide completely mineralized according to Eq. (6).

Proposal pathway of mineralization of triadimefon by PEF under experimental conditions of Fig. 3
4 Conclusion
Experimental conditions for a complete mineralization of triadimefon in water by PEF method were determined using a reticulated vitreous carbon cathode and a steel anode. Exposure of the solution at UVA radiation increases the degradation rate of triadimefon compared to the EF process. The degradation followed pseudo-first-order kinetics. 4-Chlorophenol and hydroquinone are the main aromatic products formed during the mineralization. Carboxylic acids are also produced, which in turn are further degraded under the experimental conditions used, so the fungicide in solution is transformed into CO2, water, and inorganic ions.
References
Allen, W., Wolf, D., George, M., Hester, S., Sun, G., Thai, S.-F., Delker, D., Moore, T., Jones, C., Nelson, G., Roop, B., Leavitt, S., Winkfield, E., Ward, W., & Nesnow, S. (2006). Toxicity profiles in mice treated with hepatotumorigenic and non-hepatotumorigenic triazole conazole fungicides: propiconazole, triadimefon, and myclobutanil. Toxicology Pathology, 34, 853–862.
Boye, B., Dieng, M., & Brillas, E. (2002). Degradation of herbicide 4-chlorophenoxyacetic acid by advanced electrochemical oxidation methods. Environmental Science and Technology, 36, 3030–3035.
Brillas, E., Sirés, I., & Oturan, M. (2009). Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chemical Reviews, 109, 6570–6631.
Cañizares, P., Paz, R., Lobato, J., Sáez, C., & Rodrigo, M. A. (2006). Electrochemical treatment of the effluent on a fine-chemical manufacturing plant. Journal of Hazardous Materials B, 138, 173–181.
Chang, R. (2000). Physical chemistry for the chemical and biological sciences (pp. 445–500). Sausalito: University Science Books. Chapter 12.
Chen, P.-J., Padgett, W., Moore, T., Winnik, W., Lambert, G., Thai, S.-F., Hester, S., & Nesnow, S. (2009). Three conazoles increase hepatic microsomal retinoic acid metabolism and decrease mouse hepatic retinoic acid levels in vivo. Toxicology and Applied Pharmacology, 234, 143–155.
Hu, L., Zhang, S., Li, W., & Hou, B. (2010). Electrochemical and thermodynamic investigation of diniconazole and triadimefon as corrosion inhibitors for copper in synthetic seawater. Corrosion Science, 52, 2891–2896.
Kidd, H., & James, D. R. (Eds.). (1991). The agrochemicals handbook (3rd ed.). Cambridge: Royal Society of Chemistry Information Services.
Kusic, H., Koprivanac, N., & Srsan, L. (2006). Azo dye degradation using Fenton type processes assisted by UV irradiation: a kinetic study. Journal of Photochemistry and Photobiology A: Chemistry, 181, 195–202.
Martínez-Huitle, C., & Brillas, E. (2009). Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: a general review. Applied Catalysis B: Environmental, 87, 105–145.
Peng, T.-Z., Cheng, Q., & Yang, C. F. (2001). Adsorptive behavior and electrochemical determination of the anti-fungal agent ketoconazole. Fresenius' Journal of Analytical Chemistry, 370, 1082–1086.
Poyatos, J. M., Muñio, M. M., Almecija, M. C., Torres, J. C., Hontoria, E., & Osorio, F. (2010). Advanced oxidation processes for wastewater treatment: state of the art. Water, Air, and Soil Pollution, 205, 187–204.
Quiang, Z., Chang, J.-H., & Haung, C.-P. (2002). Electrochemical generation of hydrogen peroxide from dissolved oxygen in acidic solutions. Water Research, 36, 85–94.
Ruiz, E. J., Arias, C., Brillas, E., Hernández-Ramírez, A., & Peralta-Hernández, J. M. (2011). Mineralization of Acid Yellow 36 azo dye by electro-Fenton and solar photoelectro-Fenton processes with a boron-doped diamond anode. Chemosphere, 82, 495–501.
Salazar, R., García-Segura, S., Ureta-Zañartu, M. S., & Brillas, E. (2011). Degradation of disperse azo dyes from waters by solar photoelectro-Fenton. Electrochimica Acta, 56, 6371–6379.
Sirés, I., Oturan, N., Otura, M., Rodríguez, R., Garrido, J., & Brillas, E. (2007). Electro-Fenton degradation of antimicrobials triclosan and triclocarban. Electrochimica Acta, 52, 5493–5503.
Skoumal, M., Arias, C., Cabot, P., Centellas, F., Garrido, J., Rodríguez, R., & Brillas, E. (2008). Mineralization of the biocide chloroxylenol by electrochemical advanced oxidation processes. Chemosphere, 71, 1718–1729.
Stamatis, N., Hela, D., & Konstantinou, I. (2010). Occurrence and removal of fungicides in municipal sewage treatment plant. Journal of Hazardous Materials, 175, 829–835.
Ting, W.-P., Lub, M.-C., & Huanga, Y.-H. (2008). The reactor design and comparison of Fenton, electro-Fenton and photoelectro-Fenton processes for mineralization of benzene sulfonic acid (BSA). Journal of Hazardous Materials, 156, 421–427.
Zhou, Q., Xiao, J., & Ding, Y. (2007). Sensitive determination of fungicides and prometryn in environmental water samples using multiwalled carbon nanotubes solid-phase extraction cartridge. Analytica Chimica Acta, 602, 223–228.
Acknowledgments
We are grateful to DICYT–USACh and FONDECYT Grant 11090275 for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Salazar, R., Ureta-Zañartu, M.S. Mineralization of Triadimefon Fungicide in Water by Electro-Fenton and Photo Electro-Fenton. Water Air Soil Pollut 223, 4199–4207 (2012). https://doi.org/10.1007/s11270-012-1184-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11270-012-1184-7