
Abstract
After 30 years of retroviral vector research it became clear that the parental viruses can be both friend and foe. Especially human immunodeficiency virus sparked a global pandemic, but could be converted into a versatile tool for cell therapy. For all retroviral genera, the way from virus to vector was similar resulting in split-vector systems based on the separation of the genes needed for vector particle formation and transgene expression. The first gene therapy trials, although clinically effective, revealed the genotoxicity of retroviral vectors caused by insertional mutagenesis. This issue was solved using self-inactivating vectors carrying weaker cellular promoters. Further fine-tuning was able to generate inducible systems. The current toolbox also contains vectors for the generation of induced pluripotent stem cells or efficient RNA interference. More recently the application of CRISPR-Cas9-mediated gene editing led to the development of genome-wide small guide RNA libraries targeting all human genes and single lentiviral vectors for an easy delivery of Cas9.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The goal of gene therapy is to deliver genetic material into target cells, tissues, or organs and either cure or modify the progression of a disease [1]. In addition, experimental manipulation of cells to study gene function is another important aim. Viruses are natural vehicles for genetic information since they are obligate cellular parasites [2]. Especially retroviruses are efficient tools for gene transfer due to several reasons. First, and most importantly they integrate their genetic information stably into the host cell chromosomes. This insertion is preceded by the conversion of the RNA genome into double-stranded DNA by reverse transcription [3]. Second, target cell specificity can be easily changed by incorporation of heterologous envelope proteins during vector production (so called pseudo-typing; [4]). Third, the generation of hybrid vectors including different vector designs and incorporation of elements from various viruses are feasible. In principle, all retrovirus genera can give rise to a potential gene therapy vector. So far, research focused mainly on three viruses (Fig. 1a), namely gammaretroviruses (murine leukemia virus, MLV), lentiviruses (human immunodeficiency virus, HIV), and spumaviruses (human foamy virus, HFV). Additionally, alpharetroviral vectors have been developed, because they exhibit a relative neutral integration pattern [5]. HIV-1 based vectors are widely used due to their ability to infect both dividing and non-dividing cells (Table 1) [6].

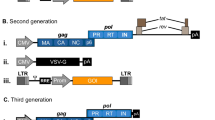
Genomes of retroviruses, the resulting vectors, and additional plasmids. a The three parental viruses for the retroviral vector types are shown. The 5′ and 3′ long terminal repeats are depicted in alternating yellow and white boxes. The color code helps to identify the core genes. Blue stands for the structural and replication genes, whereas green marks the envelope gene in all genera, respectively. In the alpha- and retroviral genome scheme, the individual protein cleavage products are named and left out for simplification in the other drawing below. For lenti- and spumaviruses the accessory genes are either colored in gray, which indicates factors involved in pathogenesis and red for essential gene expression activators. Spumaviruses as a singularity harbor an internal promoter (IP) in light violet. In addition, the top scheme marks additional sequence features common to all retroviruses such as the attachment sites for the integrase (att), the primer-binding site (PBS), the packaging signal (ψ), the 5′ splice site (SS), the polypurine tract (PPT), and the transcriptional start site (arrow below). b The vector genomes are ordered according to the scheme in a. All authentic U3 regions are replaced by the Rous sarcoma virus promoter (RSV; blue circle). All vectors are shown in the SIN vector configuration (red cross in the 3′ U3 region). Thus expression is driven by various internal promoters (IPs). The gene of interest (GOI; blue box) is followed by the woodchuck hepatitis B virus post-transcriptional regulator element (WPRE; pink box). Additional cis-acting elements including the direct repeat elements in alpharetroviral vectors (light violet) and the central PPT (gray). Lenti- and foamyviral vectors need a second element for RNA export both depicted in dark red boxes as in their parental viruses [Rev-responsive elements (RRE) and cis-acting region (CAR)]. c The paradigmatic packing plasmids are shown. These are mostly cytomegalovirus immediate-early promoter-driven expression constructs for the gag/pol and vesicular stomatitis virus glycoprotein (VSV-G) as a pantropic envelope. The Rev plasmid is only essential for lentiviral vectors, whereas gag and pol are separate plasmids for foamyviral vectors (Color figure online)
Retroviral integration is an essential step in the viral life cycle. Thus, a successful infection (transduction in the case of vectors) always leads to a stable genetic modification of the target cell [3]. This feature enables retroviral vectors to be harnessed for gene therapy, e.g., in hematopoietic stem cells (HSCs). HSCs will pass on the introduced genetic change to all progeny cells. In addition, their transduction with vector particles can be easily performed ex vivo in the absence of a functional immune system. Most of the clinical trials using retroviral vectors yielded a therapeutic effect [7], but the first generation of gammaretroviral vectors displayed a high incidence of leukemias depending on the disease background [8].
Ongoing efforts to improve or design new types of retroviral vectors significantly lowered the genotoxicity [9]. Moreover, retroviral vectors are also important tools to study heterologous envelope proteins from much more deadly viruses such as Ebola [10]. Simultaneous expression of compatible glycoproteins during vector production results in pseudo-typing of the retroviral particles [4].
In addition, recent advances resulted in retroviral vectors delivering inducible and gene editing systems, such as CRISPR-Cas9. In this review, we would like to draw a line from vector design to new applications for in vitro and in vivo studies.
From virus to vector design
The main principle of viral vector design is the identification of viral genes or elements needed for transgene delivery and deletion of the remaining sequences to generate coding capacity for the gene of interest (GOI). In a second step, the essential genes for vector production are provided on separate plasmids yielding single-round infectious viral particles. All retroviruses genera share the overall genome structure including gag, pol, and env (Fig. 1a). Gag and pol encode the structural proteins and replication enzymes, whereas env gives rise to the envelope protein anchored in the membrane enclosing the capsid. However, complex retroviruses like, e.g., lenti- and spumaviruses encode additional proteins that impact on virulence and pathogenesis (Fig. 1a; summarized in [11]).
As early as 1986 Yu et al. established the first gammaretroviral self-inactivating (SIN) vector [12]. The principle is based on the arrangement of the long terminal repeats (LTRs) flanking the viral genome 5′ and 3′. Each LTR contains a U3, R, and a U5 region. U3 represents the strong retroviral enhancer and promoter (Fig. 1a, b). During reverse transcription this part of the 3′-LTR is duplicated and transferred to the 5′ end. In SIN vectors the U3 region of the 3′-LTR is deleted and the vector inactivates itself resulting in a promoter-less vector genome. Now, the expression of the GOI must be driven by an internal promoter (Fig. 1b). This design enabled the usage for cell type-specific promoters, reduced the risk of recombination with endogenous retroviruses, and significantly lowered the probability for insertional mutagenesis [13]. The SIN vector design has been further improved in the following years to achieve high titers and robust expression of the GOI in the transduced cell [9, 14]. To do so, several cis-acting elements have been introduced. On the one hand, the woodchuck hepatitis B virus post-transcriptional regulatory element (WPRE) was incorporated into the 3′-UTR of the GOI to increase both expressions of the transgene and viral titer (Fig. 1b) [15]. On the other hand, the MLV promoter within the U3 region of the 5′-LTR has been exchanged to the Rous sarcoma virus promoter, which is especially potent in recruiting an elongation-competent RNA polymerase II yielding mostly full-length vector transcripts ready for packaging. In addition, an SV40 enhancer element recognized by the large T antigen further increased transcriptional activity in the producing cells [9]. Furthermore, the LTR is followed by the primer-binding site (PBS) and the packaging signal psi (ψ) (Fig. 1b). Interestingly, the titer of both retro- and lentiviral vectors benefits from their authentic 5′ splice site present downstream or upstream of ψ (Fig. 1b). Historically, the first generation of retroviral vectors was restricted in embryonic stem as well as embryonic carcinomas (ECs) and, more importantly, in HSCs [16]. Although it was known for many years that this was due to the sequence of the PBS and thus the choice of tRNA, the mechanism of transcriptional repression in the nucleus remained enigmatic for over a decade. Ten years ago, it has been shown that the tripartite motif containing protein 28 (TRIM28) recognizes the MLV PBS [17] and acts as a corepressor probably recruited to the DNA by different Kruppel-associated box zinc finger (KRAB-ZNF) proteins [18].
In 1996 Naldini et al. established the first generation of lentiviral vectors by deleting only the env gene [19]. Of course, the risk for the formation of replication-competent lentiviruses was not neglectable in the beginning. The Trono Laboratory enhanced the safety of the first generation lentiviral vectors by deleting all viral genes, which are dispensable for a single-round transduction (env, vif, vpr, vpu, and nef) [20]. Finally, they kept the gag, pol, tat, and rev genes. However, even the second generation of the lentiviral vectors contained overlapping sequences with the potential for recombination. The third generation of lentiviral vectors finally separated the rev gene to a plasmid while moving the Rev-responsive element (RRE) to the 5′ half of the vector genome [14]. The usage of a chimeric 5′-LTR promoter (U3 from Rous sarcoma virus, RU5 from HIV; Fig. 1b) results in Tat-independency thereby deleting another accessory gene and enhancing vector safety. Thus, a four-vector plasmid packaging system is used for lentiviral vector particle production (Fig. 1b).
Taken together, the basic vector design of retro- and lentiviral vectors is similar. Both are SIN vectors using an internal promoter for expression of the transgene. Further, the WPRE enhances the titer and GOI expression. Interestingly, the WPRE seems to compensate for the titer loss in the SIN vector configuration [9]. The basis for this reduction might be the deletion of an enhancer sequence needed for 3′ end processing of the vector RNA. This is well documented for HIV [21], but remains speculative for MLV. HIV, in contrast to simple retroviruses, contains a second initiation site for first-strand synthesis called the central polypurine tract (cPPT) [11]. Incorporation of the cPPT in lentiviral vectors presumably enhanced the completion of reverse transcription. In addition, the cPPT might be involved in nuclear import of the pre-integration complex [22].
The experiences in gamma- and lentiviral vector improvement have been transferred to foamy- and alpharetroviral vectors. The latter gained interest due to their preferentially intergenic integration pattern [23]. Despite all efforts, retroviral integration is still semi-random. This is in contrast to adeno-associated viral vectors which can be directed to specific location on chromosome 19 [24, 25].
The first clinical trials using gammaretroviral vectors for treatment of X-linked severe combined immunodeficiency (X-SCID) showed leukemic events based on the insertion of the strong retroviral enhancer/promoter elements in the vicinity of transcription start sites (TSSs) of proto-oncogenes [26]. Clonal selection after re-infusion of the cells favors ‘harmful’ integrations, if they provide a growth advantage [27, 28]. This phenomenon has been known for many years from replication-competent MLV [29]. At first this risk was neglected because the replication-incompetent vectors are only infectious for a single-round. However, clonal selection in a bone-marrow transplantation setting is a strong force quickly resulting in clonal imbalance and later in leukemic development. In the case of X-SCID vector insertion activated the LMO2 proto-oncogene thus blocking T cell differentiation leading to leukemia [26]. Lentiviral vectors favor the gene body downstream of TSS [30]. However, the usage of a strong enhancer/promoter in a lentiviral background can also activate expression of proto-oncogenes [31]. Thus, alpharetroviral vectors with their intergenic integration profile might be safer gene therapy vectors [32]. Such vectors follow the same design as other SIN vectors. Interestingly, the direct repeat elements (DREs) have to be incorporated for sufficient vector titer. DREs are RNA transport elements and may function additive to the WPRE, which is also used in alpharetroviral vectors [33, 34].
Also foamyviral SIN vectors have been designed [35]. Similar to the RRE, foamyviral vectors need an element from the central region of the genome implicated in RNA transport [36]. Transcription can be driven by the CMV promoter. One important feature of foamyviral vectors is the time point of DNA synthesis. In this respect, spumaviruses are in between retro- and hepadnaviruses (e.g., hepatitis B virus). Reverse transcription starts in the producing cell and is almost finished before entering the target cell. Thus, the nucleic acid in the vector supernatant is DNA and not RNA. In addition, gag and pol are separated expression units in the viral genome and must be supplied on two plasmids (summarized in [35].
In the following sections, we would like to mention vector designs as parts of the retroviral toolbox.
‘All-in-one’ Tet-inducible lentiviral vectors
The very well-known tetracycline (Tet)-inducible system derived from gram negative bacteria has been widely applied in molecular biology. Gossen et al. further developed the system in Tet-on and Tet-off [37]. Early lenti- and retroviral vectors could efficiently introduce a Tet-regulable promoter controlling expression of the GOI in many different tissues and cells [38, 39]. However, the regulation always relied on the pre-existing expression of the Tet-transactivator (TA) or simultaneous transduction with a vector encoding the TA. Attempts to combine both cassettes into an ‘all-in-one’ vector expressing both the TA as well as the Tet-regulated transgene were complicated by leakiness of the Tet-promoter resulting in high background expression. The uninduced Tet-promoter behaves like an enhancer-trap ‘catching’ transcriptional activity from the neighboring promoter on the vector genome [40]. Finally, Heinz et al. were able to find the correct choice and arrangement of promoters. They used the cellular phosphoglycerate kinase (PGK) promoter, which results in sufficient TA expression, but lacked transactivation potential for the nearby Tet-promoter (Fig. 2a) [41, 42]. Such vectors have been successfully used to achieve myeloprotection in anticancer chemotherapy [43].

The vector tool box: induction, reprogramming and gene editing. a The tet-regulable retroviral vector is shown. The key feature is the weak PGK promoter allowing sufficient RNA synthesis (black line) and protein expression of the reverse transactivator (M2rTA). rTA (blue circle) then activates the pTET11 promoter upon doxycycline addition. The PGK exerts only a weak cross-activation to the TET-promoter resulting in a tightly regulated all-in-one vector. Other features cf. Fig. 1b. b The reprogramming lentiviral vector uses a spleen-focus-forming-virus derived internal promoter (SF) to drive expression of a poly-protein consisting of all essential reprogramming factors. The various 2A sites allow co-translational cleavage (below the vector scheme). The marker gene GFP is expressed via an internal-ribosomal entry site (IRES, violet box). c A lentiviral vector used for gene silencing via short hairpin (sh) RNAs is depicted. The shRNA (stem loop structure) is expressed under control of the U6 promoter. A puromycin resistance gene can be used for selection. d The Cas9 lentiviral vector expresses the Cas9 protein driven by an internal promoter (IP; orange box) and a sgRNA directed by a U6 promoter (yellow box). The single-stranded sgRNA is shown below. GFP is incorporated via a P2A sequence as in 2B. All these vector systems are available through the Addgene plasmid repository using e.g., the keywords “shRNA lentiviral vectors” or “CRISPR-Cas9 lentiviral vectors” (Color figure online)
Reprogramming lentiviral vectors
The generation of induced pluripotent stem cells (iPSCs) strictly depends on the expression of the reprogramming factors (RFs) Oct4, KIF4, Sox2, c-Myc [44]. So far, different gene delivery technologies have been tested for RF transfer into murine and human somatic cells. The first studies have been performed using gammaretroviral vectors encoding every factor separately, but the efficacy of reprogramming was low [44]. In addition, several transductions result in multiple integrations, possibly promoting insertional mutagenesis. Hence, the aim was to generate a polycistronic lentiviral vector co-expressing all RFs. In the end, all RF ORFs are separated by different co-translational 2A-proteinase sites (Fig. 2b) [45]. To monitor and understand early stages in cellular reprogramming, the Schambach Laboratory improved RF vector design and introduced a fluorescent marker [46]. To temporally restrict RF expression a retroviral promoter was used, which is silenced in iPSCs during the process of reprogramming. In general, all vector systems leading to integration of the GOI will elicit side effects especially in highly proliferative iPSCs. Novel vector designs aim to overcome this drawback by removal of the vector after successful reprogramming. One idea is a Flp recombinase (Flp)-excisable lentiviral reprogramming vector. Excision leaves only 300 bp residual LTR sequence in the genome, which can also function as a DNA barcode. Further, in this reprogramming locus, an additional transgene of interest can be introduced by flp recombinase-mediated cassette exchange [47]. Another approach to prevent insertional mutagenesis is episomal transfer of RFs utilizing integration-deficient lentiviral vectors (IDLVs; [48]). The viral DNA is not integrated due to an integrase mutation and is converted into 1- and 2-LTR circles by the cellular DNA repair. These circles provide transient RF expression sufficient for iPS generation from human fibroblasts and vanish after ongoing cell division [49].
RNA interference mediated by retroviral vectors
Retroviral vectors have not only been used for gene delivery, but also for the manipulation of host gene expression by stable RNA interference (RNAi). Short hairpin (sh)RNAs are introduced via retroviral vectors to knock down the expression of a gene in vitro and in vivo [50, 51]. shRNA expression cassettes under control of a pol-III promoter (e.g., H1 or U6) are usually inserted downstream of the RRE in lentiviral vectors (Fig. 1; [52]). An interesting variation is cloning of the shRNA into the U3 deletion in the SIN vector configuration. This design results in a duplication after integration thereby increasing efficacy [51]. Another versatile tool to perform RNAi is the utilization of microRNAs (miRNAs). miRNAs are small, cellular non-coding RNAs, exhibiting differential expression in various cell types and tissues [53]. Many of them are highly conserved between species. In contrast to siRNA and shRNAs, which result in cleavage of target mRNAs, miRNAs inhibit translation and may finally result in accelerated RNA degradation [53]. The hallmark of miRNAs is a partial, non-perfect homology to the target sequence. This property allows them to target many mRNAs at the same time. miRNAs are transcribed by a RNA pol-II resulting in primary-polyadenylated RNA (pri-miRNA). The pri-miRNA is further cleaved by the enzyme Drosha yielding a pre-miRNA of about 70–90 nucleotides (nts), which is exported to the cytoplasm [54]. Finally, the enzyme Dicer creates the mature ~22-nts-long miRNA as it does for shRNAs. Since miRNAs are pol-II-derived transcripts, regulated expression via a cell specific or regulable (e.g., Tet) promoter is possible (reviewed in [55]). Retro- and lentiviral vectors encoding miRNAs can be used to control transgene expression itself or to downregulate cellular or viral gene expression in a therapeutic approach. Likewise, the incorporation of miRNA target sites can result in cell type-specific expression. The twist is that if target sites for liver-specific miRNAs are used, expression in hepatocytes is restricted. This can be important if the transgene exerts negative effect in one cell type but not the other. For suppression of transgene expression, multiple copies of the miRNA sequence have to be placed in the 3′-UTR of the GOI. Using a bidirectional lentiviral vector expressing a marker gene in sense direction and miRNA target site placed in the 3′-UTR of the GOI in antisense direction, Brown et al. were able to achieve lineage-specific expression of the GOI [56]. Finally, miRNA delivering lentiviral vectors are suitable tools to generate stable knockdown cell lines and transgenic animals [57].
CRISPR-Cas9 vectors
As soon as gene editing by CRISPR-Cas9 was shown to function in human cells, all-in-one lentiviral vectors expressing both Cas9 and single guide RNAs (sgRNAs) were designed (Fig. 2c; [58]). In principle, they follow the same structure as vectors expressing shRNAs and a marker/selection gene. As with shRNAs, libraries consisting of thousands of sgRNAs have been constructed. Such CRISPR libraries have been used in a genome-wide loss of function screen. In this study, the authors used sgRNAs targeting 5′ constitutive exons and show potent gene knockout [59]. In a more functional approach, a CRISPR screen using lentiviral Cas9 vectors identified novel tumor suppressor genes involved in acute leukemia [60]. On a smaller, array-type scale overlapping sgRNAs have been designed and pooled to allow mutational analysis of key sequence elements directing the switch from fetal to adult hemoglobin [61]. DNA double-strand breaks introduced by CRISPR-Cas9 can also enhance homology-directed gene repair (HDR). In current approaches, the donor template for HDR is expressed from IDLVs on a separate vector genome [62]. With the discovery of Cas9 off-target sites, temporal restriction of Cas9 expression becomes more important. Here, the all-in-one Tet vectors might be of special interest.
In the future, CRISPR Cas9-mediated gene editing will be used to manipulate the genome either for gene knockout or HDR. Lentiviral vectors are theoretically capable of providing the Cas9, sgRNA, and a repair template in a single vector. This would raise the possibility to restore, e.g., deletions or point mutations causing diseases. Very recently, the first Cas9-edited cells have been re-infused into cancer patients (doi: 10.1038/nature.2016.20988).
Summary
Taken together, retro- and lentiviral vectors have been very successfully used for treating monogenetic diseases [63]. Although the first and second generation of retroviral vectors suffered from enhanced genotoxicity, the third SIN vector generation using weaker cellular promoters did not show any side effects [64]. Most clinical trials are now performed with lentiviral vectors targeting mostly disorders of the hematopoietic system including chimeric antigen receptor (CAR) anticancer therapy [65]. Still some drawbacks exist such as restricted temporal or spatial expression of the GOI. As mentioned above, the incorporation of miRNA-binding sites is one approach to solve this problem. However, gain-of-function mutations will never be cured using lentiviral additive gene therapy. In the foreseeable future, gene repair using genome editing techniques such as CRISPR-Cas9 will continue to rise.
References
R.W. Herzog, O. Cao, A. Srivastava, Discov. Med. 9, 105–111 (2010)
S.J. Flint, L.W. Enquist, V.R. Racaniello, A.M. Skalka, Principles of Virology (ASM Press, Washington, DC, 2009)
H.E. Varmus, Science 216, 812–820 (1982)
S.J. Russell, F.L. Cosset, J. Gene Med. 1, 300–311 (1999)
J.D. Suerth, T. Maetzig, M. Galla, C. Baum, A. Schambach, J. Virol. 84, 6626–6635 (2010)
L. Naldini, D. Trono, I.M. Verma, Science 353, 1101–1102 (2016)
B.L. Alexander, R.R. Ali, E.W. Alton, J.W. Bainbridge, S. Braun, S.H. Cheng, T.R. Flotte, H.B. Gaspar, M. Grez, U. Griesenbach, M.G. Kaplitt, M.G. Ott, R. Seger, M. Simons, A.J. Thrasher, A.Z. Thrasher, S. Yla-Herttuala, Gene Ther. 14, 1439–1447 (2007)
S. Hacein-Bey-Abina, C. von Kalle, M. Schmidt, F. Le Deist, N. Wulffraat, E. McIntyre, I. Radford, J.L. Villeval, C.C. Fraser, M. Cavazzana-Calvo, A. Fischer, N. Engl. J. Med. 348, 255–256 (2003)
A. Schambach, D. Mueller, M. Galla, M.M. Verstegen, G. Wagemaker, R. Loew, C. Baum, J. Bohne, Gene Ther. 13, 1524–1533 (2006)
G.P. Kobinger, D.J. Weiner, Q.C. Yu, J.M. Wilson, Nat. Biotechnol. 19, 225–230 (2001)
J.M. Coffin, S.H. Hughes, H.E. Varmus, Retroviruses (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1997)
S.F. Yu, T. von Ruden, P.W. Kantoff, C. Garber, M. Seiberg, U. Ruther, W.F. Anderson, E.F. Wagner, E. Gilboa, Proc. Natl Acad. Sci. USA 83, 3194–3198 (1986)
U. Modlich, J. Bohne, M. Schmidt, C. von Kalle, S. Knob, A. Schambach, C. Baum, Blood 108, 2545–2553 (2006)
T. Dull, R. Zufferey, M. Kelly, R.J. Mandel, M. Nguyen, D. Trono, L. Naldini, J. Virol. 72, 8463–8471 (1998)
R. Zufferey, J.E. Donello, D. Trono, T.J. Hope, J. Virol. 73, 2886–2892 (1999)
M. Grez, E. Akgun, F. Hilberg, W. Ostertag, Proc. Natl Acad. Sci. USA 87, 9202–9206 (1990)
D. Wolf, S.P. Goff, Cell 131, 46–57 (2007)
R. Urrutia, Genome Biol. 4, 231 (2003)
L. Naldini, U. Blomer, P. Gallay, D. Ory, R. Mulligan, F.H. Gage, I.M. Verma, D. Trono, Science 272, 263–267 (1996)
R. Zufferey, D. Nagy, R.J. Mandel, L. Naldini, D. Trono, Nat. Biotechnol. 15, 871–875 (1997)
A. Furger, J. Monks, N.J. Proudfoot, J. Virol. 75, 11735–11746 (2001)
V. Zennou, C. Petit, D. Guetard, U. Nerhbass, L. Montagnier, P. Charneau, Cell 101, 173–185 (2000)
A. Narezkina, K.D. Taganov, S. Litwin, R. Stoyanova, J. Hayashi, C. Seeger, A.M. Skalka, R.A. Katz, J. Virol. 78, 11656–11663 (2004)
R.M. Kotin, M. Siniscalco, R.J. Samulski, X.D. Zhu, L. Hunter, C.A. Laughlin, S. McLaughlin, N. Muzyczka, M. Rocchi, K.I. Berns, Proc. Natl Acad. Sci. USA 87, 2211–2215 (1990)
R.J. Samulski, X. Zhu, X. Xiao, J.D. Brook, D.E. Housman, N. Epstein, L.A. Hunter, EMBO J. 10, 3941–3950 (1991)
S. Hacein-Bey-Abina, C. Von Kalle, M. Schmidt, M.P. McCormack, N. Wulffraat, P. Leboulch, A. Lim, C.S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P. Fraser, J.I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A. Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E. Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L.E. Leiva, M. Wissler, C. Prinz, T.H. Rabbitts, F. Le Deist, A. Fischer, M. Cavazzana-Calvo, Science 302, 415–419 (2003)
H. Glimm, M. Schmidt, M. Fischer, K. Schwarzwaelder, M. Wissler, S. Klingenberg, C. Prinz, C.F. Waller, W. Lange, C.J. Eaves, C. von Kalle, Blood 106, 893–898 (2005)
M.G. Ott, M. Schmidt, K. Schwarzwaelder, S. Stein, U. Siler, U. Koehl, H. Glimm, K. Kuhlcke, A. Schilz, H. Kunkel, S. Naundorf, A. Brinkmann, A. Deichmann, M. Fischer, C. Ball, I. Pilz, C. Dunbar, Y. Du, N.A. Jenkins, N.G. Copeland, U. Luthi, M. Hassan, A.J. Thrasher, D. Hoelzer, C. von Kalle, R. Seger, M. Grez, Nat. Med. 12, 401–409 (2006)
L.F. Lock, N.A. Jenkins, N.G. Copeland, Curr. Top. Microbiol. Immunol. 171, 27–41 (1991)
R.S. Mitchell, B.F. Beitzel, A.R. Schroder, P. Shinn, H. Chen, C.C. Berry, J.R. Ecker, F.D. Bushman, PLoS Biol. 2, E234 (2004)
E. Montini, D. Cesana, M. Schmidt, F. Sanvito, C.C. Bartholomae, M. Ranzani, F. Benedicenti, L.S. Sergi, A. Ambrosi, M. Ponzoni, C. Doglioni, C. Di Serio, C. von Kalle, L. Naldini, J. Clin. Investig. 119, 964–975 (2009)
J.D. Suerth, T. Maetzig, M.H. Brugman, N. Heinz, J.U. Appelt, K.B. Kaufmann, M. Schmidt, M. Grez, U. Modlich, C. Baum, A. Schambach, Mol. Ther. 20, 1022–1032 (2012)
R.A. Ogert, L.H. Lee, K.L. Beemon, J. Virol. 70, 3834–3843 (1996)
J. Sorge, W. Ricci, S.H. Hughes, J. Virol. 48, 667–675 (1983)
D. Lindemann, A. Rethwilm, Viruses 3, 561–585 (2011)
H. Wodrich, J. Bohne, E. Gumz, R. Welker, H.G. Krausslich, J. Virol. 75, 10670–10682 (2001)
M. Gossen, H. Bujard, Proc. Natl Acad. Sci. USA 89, 5547–5551 (1992)
J.K. Koponen, H. Kankkonen, J. Kannasto, T. Wirth, W. Hillen, H. Bujard, S. Yla-Herttuala, Gene Ther. 10, 459–466 (2003)
D.S. Vieyra, M.A. Goodell, Stem Cells 25, 2559–2566 (2007)
E. Vigna, M. Amendola, F. Benedicenti, A.D. Simmons, A. Follenzi, L. Naldini, Mol. Ther. 11, 763–775 (2005)
R. Loew, N. Heinz, M. Hampf, H. Bujard, M. Gossen, BMC Biotechnol. 10, 81 (2010)
N. Heinz, A. Schambach, M. Galla, T. Maetzig, C. Baum, R. Loew, B. Schiedlmeier, Hum. Gene Ther. 22, 166–176 (2011)
N. Lachmann, S. Brennig, R. Hillje, H. Schermeier, R. Phaltane, J. Dahlmann, I. Gruh, N. Heinz, B. Schiedlmeier, C. Baum, T. Moritz, Gene Ther. 22, 883–892 (2015)
K. Takahashi, S. Yamanaka, Cell 126, 663–676 (2006)
B.W. Carey, S. Markoulaki, J. Hanna, K. Saha, Q. Gao, M. Mitalipova, R. Jaenisch, Proc. Natl Acad. Sci. USA 106, 157–162 (2009)
E. Warlich, J. Kuehle, T. Cantz, M.H. Brugman, T. Maetzig, M. Galla, A.A. Filipczyk, S. Halle, H. Klump, H.R. Scholer, C. Baum, T. Schroeder, A. Schambach, Mol. Ther. 19, 782–789 (2011)
J. Kuehle, S. Turan, T. Cantz, D. Hoffmann, J.D. Suerth, T. Maetzig, D. Zychlinski, C. Klein, D. Steinemann, C. Baum, J. Bode, A. Schambach, Mol. Ther. 22, 919–928 (2014)
A. Lombardo, P. Genovese, C.M. Beausejour, S. Colleoni, Y.L. Lee, K.A. Kim, D. Ando, F.D. Urnov, C. Galli, P.D. Gregory, M.C. Holmes, L. Naldini, Nat. Biotechnol. 25, 1298–1306 (2007)
J.W. Schott, D. Hoffmann, T. Maetzig, F.J. Muller, D. Steinemann, D. Zychlinski, T. Cantz, C. Baum, A. Schambach, Gene Ther. 21, 938–949 (2014)
E. Devroe, P.A. Silver, BMC Biotechnol. 2, 15 (2002)
G. Tiscornia, O. Singer, M. Ikawa, I.M. Verma, Proc. Natl Acad. Sci. USA 100, 1844–1848 (2003)
J. Moffat, D.A. Grueneberg, X. Yang, S.Y. Kim, A.M. Kloepfer, G. Hinkle, B. Piqani, T.M. Eisenhaure, B. Luo, J.K. Grenier, A.E. Carpenter, S.Y. Foo, S.A. Stewart, B.R. Stockwell, N. Hacohen, W.C. Hahn, E.S. Lander, D.M. Sabatini, D.E. Root, Cell 124, 1283–1298 (2006)
D.P. Bartel, Cell 136, 215–233 (2009)
R.L. Skalsky, B.R. Cullen, Annu. Rev. Microbiol. 64, 123–141 (2010)
B. Gentner, L. Naldini, Tissue Antigens 80, 393–403 (2012)
B.D. Brown, M.A. Venneri, A. Zingale, L. Sergi Sergi, L. Naldini, Nat. Med. 12, 585–591 (2006)
B. Gentner, G. Schira, A. Giustacchini, M. Amendola, B.D. Brown, M. Ponzoni, L. Naldini, Nat. Methods 6, 63–66 (2009)
A. Malina, J.R. Mills, R. Cencic, Y. Yan, J. Fraser, L.M. Schippers, M. Paquet, J. Dostie, J. Pelletier, Genes Dev. 27, 2602–2614 (2013)
O. Shalem, N.E. Sanjana, E. Hartenian, X. Shi, D.A. Scott, T.S. Mikkelsen, D. Heckl, B.L. Ebert, D.E. Root, J.G. Doench, F. Zhang, Science 343, 84–87 (2014)
D. Heckl, M.S. Kowalczyk, D. Yudovich, R. Belizaire, R.V. Puram, M.E. McConkey, A. Thielke, J.C. Aster, A. Regev, B.L. Ebert, Nat. Biotechnol. 32, 941–946 (2014)
M.C. Canver, E.C. Smith, F. Sher, L. Pinello, N.E. Sanjana, O. Shalem, D.D. Chen, P.G. Schupp, D.S. Vinjamur, S.P. Garcia, S. Luc, R. Kurita, Y. Nakamura, Y. Fujiwara, T. Maeda, G.C. Yuan, F. Zhang, S.H. Orkin, D.E. Bauer, Nature 527, 192–197 (2015)
I. Maggio, M.A. Goncalves, Trends Biotechnol. 33, 280–291 (2015)
A. Aiuti, L. Biasco, S. Scaramuzza, F. Ferrua, M.P. Cicalese, C. Baricordi, F. Dionisio, A. Calabria, S. Giannelli, M.C. Castiello, M. Bosticardo, C. Evangelio, A. Assanelli, M. Casiraghi, S. Di Nunzio, L. Callegaro, C. Benati, P. Rizzardi, D. Pellin, C. Di Serio, M. Schmidt, C. Von Kalle, J. Gardner, N. Mehta, V. Neduva, D.J. Dow, A. Galy, R. Miniero, A. Finocchi, A. Metin, P.P. Banerjee, J.S. Orange, S. Galimberti, M.G. Valsecchi, A. Biffi, E. Montini, A. Villa, F. Ciceri, M.G. Roncarolo, L. Naldini, Science 341, 1233151 (2013)
S. Hacein-Bey-Abina, S.Y. Pai, H.B. Gaspar, M. Armant, C.C. Berry, S. Blanche, J. Bleesing, J. Blondeau, H. de Boer, K.F. Buckland, L. Caccavelli, G. Cros, S. De Oliveira, K.S. Fernandez, D. Guo, C.E. Harris, G. Hopkins, L.E. Lehmann, A. Lim, W.B. London, J.C. van der Loo, N. Malani, F. Male, P. Malik, M.A. Marinovic, A.M. McNicol, D. Moshous, B. Neven, M. Oleastro, C. Picard, J. Ritz, C. Rivat, A. Schambach, K.L. Shaw, E.A. Sherman, L.E. Silberstein, E. Six, F. Touzot, A. Tsytsykova, J. Xu-Bayford, C. Baum, F.D. Bushman, A. Fischer, D.B. Kohn, A.H. Filipovich, L.D. Notarangelo, M. Cavazzana, D.A. Williams, A.J. Thrasher, N. Engl. J. Med. 371, 1407–1417 (2014)
A.D. Posey Jr., R.D. Schwab, A.C. Boesteanu, C. Steentoft, U. Mandel, B. Engels, J.D. Stone, T.D. Madsen, K. Schreiber, K.M. Haines, A.P. Cogdill, T.J. Chen, D. Song, J. Scholler, D.M. Kranz, M.D. Feldman, R. Young, B. Keith, H. Schreiber, H. Clausen, L.A. Johnson, C.H. June, Immunity 44, 1444–1454 (2016)
Acknowledgements
We would like to thank V. Melhorn for providing initial art work. This review was partially funded by the Else-Kröner-Fresenius-Stiftung given to J. B. (2014_A88).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Anja Ehrhardt and Florian Kreppel
Rights and permissions
About this article

Cite this article
Elsner, C., Bohne, J. The retroviral vector family: something for everyone. Virus Genes 53, 714–722 (2017). https://doi.org/10.1007/s11262-017-1489-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-017-1489-0