
Abstract
Dengue has affected Indonesia for the last five decades and become a major health problem in many cities in the country. Jakarta, the capital of Indonesia, reports dengue cases annually, with several outbreaks documented. To gain information on the dynamic and evolutionary history of dengue virus (DENV) in Jakarta, we conducted phylogenetic and evolutionary analyses of DENV isolated in 2009. Three hundred thirty-three dengue-suspected patients were recruited. Our data revealed that dengue predominantly affected young adults, and the majority of cases were due to secondary infection. A total of 171 virus isolates were successfully serotyped. All four DENV serotypes were circulating in the city, and DENV-1 was the predominant serotype. The DENV genotyping of 17 isolates revealed the presence of Genotypes I and IV in DENV-1, while DENV-2 isolates were grouped into the Cosmopolitan genotype. The grouping of isolates into Genotype I and II was seen for DENV-3 and DENV-4, respectively. Evolutionary analysis revealed the relatedness of Jakarta isolates with other isolates from other cities in Indonesia and isolates from imported cases in other countries. We revealed the endemicity of DENV and the role of Jakarta as the potential source of imported dengue cases in other countries. Our study provides genetic information regarding DENV from Jakarta, which will be useful for upstream applications, such as the study of DENV epidemiology and evolution and transmission dynamics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dengue disease is a systemic viral infection caused by dengue virus (DENV), a member of the Flaviviridae family. It is estimated that 390 million cases of dengue infection occur annually, of which 96 million manifest apparently [1]. The clinical manifestations of dengue range from the mild dengue fever (DF) to the more severe forms of the disease, dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) [2]. The DENV genome consists of a ~10.7-kb single-stranded positive-sense RNA that encodes three structural (C, prM/M, E) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) proteins [3]. DENV has diverse genetic characteristics, as reflected by the presence of four serotypes [4]. Each serotype of DENV further harbors extensive genetic diversity in the form of phylogenetically distinct clusters termed genotypes. These genotypes differ in their geographical distributions, fitness, and virulence [5, 6].
Genetic analysis of DENV is important to supplement epidemiological data with information that can be used to reconstruct the history of epidemics in time and space [7]. Phylogenetic analysis of viral genomic sequences can be used to understand DENV evolution and its effects on virus transmission and disease. DENV genotype identification can be performed using coding genes, with the Envelope (E) gene as the frequently selected target [8].
Dengue is hyperendemic in Indonesia, with all four serotypes of DENV circulating in the region [9]. Frequent epidemic cycles have occurred, and currently all provinces in Indonesia have reported dengue cases [10]. Major dengue outbreaks have been reported in 1973, 1988, 1998, 2007, and 2010 [11], with some reports on the virological aspects of the outbreaks, such as those in 1998 in Palembang, South Sumatra [12] and in 2004 in Jakarta [13, 14]. Complex urban settings with a very dense population have aided the dengue transmission and outbreak potential in this city [14]. However, limited data on the virological and epidemiological aspects of dengue in this city may hinder case management and outbreak preparedness.
The documentation of DENV serotype spread has important implications for the understanding of some key aspects of dengue infection, such as patterns in dengue hyperendemicity, disease severity, vaccine design, and deployment strategies. Jakarta is one of the sites for dengue vaccine clinical trials [15]. Information on the circulation of DENV serotypes and genotypes in Jakarta will be useful since vaccine efficacy could depend on the similarity of the DENV genotypes used in vaccine production and those circulating in the area of vaccine introduction [16]. Dengue disease investigation in an outbreak area will be particularly useful for understanding the etiology of the disease, the DENV serotype most responsible for the outbreak, the introduction of novel serotypes/genotypes in locations where DENV is or is not already present, and DENV population structure, and evolution [9]. To gain information on the dynamic and evolutionary history of dengue in Jakarta, we conducted phylogenetic and evolutionary analyses on DENV isolated in 2009.
Materials and methods
Study site, patient recruitment, and sample collection
This cross-sectional study was conducted in Jakarta, the capital city of Indonesia and the largest city in the country. Involvement of human subjects in this study was approved by the National Institute for Health Research and Development (NIHRD) Research Ethics Commission number LB.03.02/KE/4537/2009, June 24th 2009. Dengue-suspect patients were recruited from three hospitals, RS Koja, RS Budi Asih, and RS Tarakan, during the dengue peak season of September 2009 through February 2010. Written informed consents were obtained from the patients or their legal guardian. The inclusion criteria were patients with a fever >38 °C accompanied by at least one of the clinical signs of dengue, such as malaise, arthralgia, rash, retro-orbital pain, or signs of DHF or DSS. Venous blood samples were obtained from a total of 333 patients. The demographic and clinical data of the patients were recorded.
Dengue diagnosis, detection, and serotyping
Dengue was diagnosed by detection of the NS1 antigen using the Panbio Dengue Early ELISA kit (Alere, Brisbane, Australia). Serological testing and the determination of primary versus secondary infection were performed using the Panbio Dengue Duo IgM and IgG ELISA (Alere) according to the manufacturer’s protocol.
Virus RNA was extracted from 140 µL of serum samples or culture supernatant using the QIAamp Viral RNA Mini kit (Qiagen, Germany) according to the manufacturer’s protocol. The detection of DENV RNA in samples and the subsequent serotype determination were performed initially using the two-step RT-PCR method using the protocol described by Lanciotti, et al. [17] with modifications according to Harris, et al. [18]. The results from this conventional method were reconfirmed using a Simplexa Dengue real-time RT-PCR assay (Focus Diagnostics, Cypress, CA) [19].
Virus isolation
Serum samples identified as dengue and confirmed by NS1 and/or RT-PCR were then subjected to a maximum of two passages of virus isolation in cell culture. A total of 200 µL of serum was inoculated into a monolayer C6/36 (Aedes albopictus, mid gut) cell line in 2 mL of 1 × RPMI medium supplemented with 2% Fetal Bovine Serum (FBS) (Gibco-Thermo Scientific, USA). Following a virus adsorption period at 28 °C for 1 h, the inoculation medium was discarded and replenished with 3 mL of fresh medium. Infected cells were incubated at 28 °C until the cytopathic effect (CPE) was identified or up to 9 days for initial harvest, continuing with an additional 5 days where CPE was not detected. Supernatant was clarified by centrifugation at 4000 rpm (1520 × g).
DENV genotyping
Genotyping was performed based on the Envelope (E) gene. DENV RNA was reverse-transcribed into cDNA using Superscript III Reverse Transcriptase (Invitrogen-Thermo Scientific) and then PCR-amplified using Pfu Turbo Polymerase (Stratagene-Agilent Technologies, USA). PCR products were purified from 0.8% agarose gel using the QIAquick gel extraction kit (Qiagen), and cycle sequencing reactions were performed with 6 overlapping primers for each serotype from both strands (Supplementary Table) and BigDye Dideoxy Terminator sequencing kits v.3.1 (Applied Biosystems-Thermo Scientific). Purified DNA underwent capillary sequencing performed on a 3130xl Genetic Analyzer (Applied Biosystems). Sequence reads were assembled using SeqScape v.2.5 software (Applied Biosystems) with manual inspection performed when ambiguities were present. Contigs were generated and used in subsequent analyses.
DENV phylogenetic and evolutionary analyses
To analyze the phylogenetic and evolutionary information and the relatedness of Jakarta isolates with other isolates worldwide, E gene sequences of Jakarta 2009 DENV isolates were aligned together with all publicly available DENV sequences in GenBank as of 1 April 2016. Retrieved sequences from each serotype were screened to remove all nonrelated sequences or coding sequences of single genes other than E gene. The initial screening yielded taxa numbers of 4143, 1318, 2004, and 1378, for DENV-1, -2, -3, and -4, respectively.
Multiple sequence alignment was performed using MAFFT software [20]. The resulting alignment of 1485 nt (1479 nt for DENV-3) was then used to generate an initial UPGMA tree based on genetic distance. To clarify the tree, we then selected strains with a known isolation year that were most closely related to our Jakarta isolates to generate a set of 60 taxons per serotype. The trimming and pruning were done using Jalview desktop 2.9 software [21]. The possibility of recombination events in each dataset was analyzed using RDP3 software [22].
Robust phylogenetic and evolutionary analyses were then performed on the sequences. The dataset for each serotype was prepared using the BEAUti v.1.8.2 graphical interface, with the tip of each isolate calibrated using the year of isolation. Phylogenetic reconstruction and evolutionary rate analysis were obtained using the Bayesian Markov Chain Monte Carlo (MCMC) method, as implemented in BEAST v.1.8.2. The phylogenetic tree was inferred based on the selection of the statistical model for likelihood calculation optimized for the Maximum Likelihood (ML) tree using jModelTest v.2.1.4 [23]. Phylogenetic reconstruction was performed using the General Time Reversible (GTR) model with four Gamma parameters (G4) and invariant (I) sites, a relaxed uncorrelated lognormal molecular clock and Bayesian skyline prior, with 100 million generations and sampling for every 1000th iteration, and employing a 10% burn-in. The initial estimated evolutionary rate was set at 7.6 × 10−4 substitutions per site per year, as previously described [7]. The MCMC trace was analyzed using Tracer v.1.5.0 to monitor adequate Effective Sampling Size (ESS) for all parameters. A maximum clade credibility (MCC) tree was created using TreeAnnotator v.1.8.2 and visualized using FigTree v.1.4.0. The evolutionary parameters were estimated as the median number with 95% highest posterior density (HPD). The classification of genotypes in each serotype was based on classifications by Goncalvez et al. [24]., Twiddy et al. [25], Lanciotti et al. [26], and Lanciotti et al. [27] for DENV-1, -2, -3 and -4, respectively. E gene sequences similarity analysis was performed using SIAS program (http://imed.med.ucm.es/Tools/sias.html).
Results
Dengue diagnosis, incidence, patient demographics, and serotype distribution
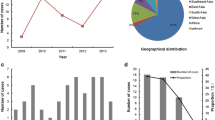
Serum samples from 333 dengue-suspect patients were collected from September 2009 through February 2010. The age distribution of the patients was from 6 months to 72 years old. Young adults (age 11–20 years) were the predominant age group (Fig. 1a). In terms of infection status, the majority (81.3%) of patients had secondary infections. A total of 171 samples (51.4%) were confirmed to have dengue infection using RT-PCR and/or NS1 antigen detection. DENV-1 was the most predominant serotype circulating in the city, found in 37.4% of samples. This percentage was followed by DENV-3 (24.0%), DENV-2 (21.1%), and DENV-4 (14.0%). Six samples (3.5%) were identified as mixed infections of two different serotypes (Fig. 1b). Among all samples that were successfully serotyped, NS1 detection was positive in 68 (39.8%) samples; with the lowest detection was observed in DENV-4 samples (29.2%). In terms of correlation between infection status (primary vs secondary infection) and DENV serotypes, we did not observe any statistically significant correlation (p = 0.7951, data not shown).

The distribution of dengue cases in Jakarta in 2009 determined by the age of dengue-suspect patients (a), and the circulating virus serotypes (b)
DENV genotype distribution and evolutionary analyses
Looking deeper into the genotypes within the serotypes, we performed genotyping based on the E gene. The genotyping approach used RNA extracted from culture supernatant as the RNA template source. Out of 171 confirmed dengue samples, 33 (19.3%) viruses were successfully isolated in cell culture. The full-length E gene was generated for 17 representative isolates, including all four serotypes. Isolate sequences were deposited into GenBank with granted accession numbers from KX646375 to KX646391.
Within the DENV-1 serotype, we observed the presence of two different genotypes. Based on Goncalvez classification [24], of seven DENV-1 isolates, six isolates (85.7%) were grouped into Genotype I, and one isolate (14.3%) was grouped into Genotype IV (Fig. 2). Within the Genotype I isolates, the DENV-1 Jakarta isolates were closely related to strains from imported cases in Taiwan in 2009 and 2010 [28] and an imported case in Western Australia in 2010 [29]. A relatedness of Jakarta isolates with strains from Sukabumi [30] and Singapore in 2009 [31] was also observed. One isolate of DENV-1 Genotype IV was grouped together in a clade including strains from imported cases in Taiwan and Japan, as well as from Malaysia and the islands of the Pacific (Reunion and Seychelles Islands). We also observed relatedness of the Jakarta isolate to local strains from the city of Bogor, West Java, which is located approximately 50 km south of Jakarta [32], and a strain from a 2010 Dengue study in Jakarta [33]. The mean evolutionary rate of DENV-1 as calculated by BEAST was 9.93 × 10−4 subs/site/year [95% HPD 6.55–14.33 × 10−4].

Phylogenetic tree of DENV-1 isolates from Jakarta (red labels) and their most closely related reference sequences from GenBank repository. The DENV-1 Jakarta isolates were classified into Genotype I and IV. Tree was generated by the Bayesian inference method as implemented in BEAST using the GTR + G4 + I evolution model calculated using E gene sequences. The number in the node indicates the posterior probability of that particular cluster, with values higher than 0.5 shown (Color figure online)
Four DENV-2 isolates were grouped into the Cosmopolitan genotype, according to the Twiddy classification [25]. Among them, three isolates were closely related and grouped into a clade with strains from a previous outbreak in Jakarta in 2004 [14] and one strain from concurrent dengue infections in Jakarta in 2013 [34]. One isolate showed a close relationship to strains from Guangdong, China (Fig. 3). Using the Bayesian algorithm, the estimated mean evolutionary rate of DENV-2 was 11.92 × 10−4 subs/site/year [95% HPD 8.70–15.89 × 10−4].

Phylogenetic tree of DENV-2 isolates from Jakarta (red labels) and their most closely related reference sequences from GenBank repository. The DENV-2 Jakarta isolates were classified into Cosmopolitan Genotype. Tree was generated by the Bayesian inference method as implemented in BEAST using the GTR + G4 + I evolution model calculated using E gene sequences. The number in the node indicates the posterior probability of that particular cluster, with values higher than 0.5 shown (Color figure online)
We successfully genotyped three isolates of DENV-3 from Jakarta, which were grouped into Genotype I according to Lanciotti classification [26]. Figure 4 shows that the Jakarta isolates were clustered into two distinct clades with high posterior support. The Jakarta isolate T-007 was grouped together with strains from Singapore [31] and Surabaya, East Java, Indonesia [35]. The other isolate, T-094, was grouped together with strains from imported cases in Taiwan [28] and Australia (from Bali) [36]. One isolate (K-136) was grouped together with Jakarta strains from the previous 2004 [14] and 2010 [33] epidemics. The mean evolutionary rate of DENV-3 was 9.32 × 10−4 subs/site/year [95% HPD 7.56–11.26 × 10−4].

Phylogenetic tree of DENV-3 isolates from Jakarta (red labels) and their most closely related reference sequences from GenBank repository. The DENV-3 Jakarta isolates were classified into Genotype I. Tree was generated by the Bayesian inference method as implemented in BEAST using the GTR + G4 + I evolution model calculated using E gene sequences. The number in the node indicates the posterior probability of that particular cluster, with values higher than 0.5 shown (Color figure online)
For DENV-4, we managed to genotype 3 isolates, which were grouped into Genotype II according to Lanciotti classification [27]. Two isolates were clustered together with isolates of imported cases in Taiwan [28, 37]. The other isolate was grouped together with and closely related to strains from Indonesia [27]. Utilizing BEAST calculations, the DENV-4 mean evolutionary rate was 9.96 × 10−4 subs/site/year [95% HPD 6.89–13.18 × 10−4].
Discussion
Indonesia has become a dengue hyperendemic country where there have been multiple epidemic cycles, and the spread of all four serotypes of DENV has reached all of the provinces of Indonesia [10]. Currently, only limited DENV genetic information is available in Indonesia, although data were available for some cities. Information on the genetic characteristics of the responsible DENV serotypes may aid in the better management of dengue. As the capital of Indonesia, Jakarta has been the melting pot of the nation and the center of globalization and urbanization from other cities in Indonesia. These conditions provides a fertile ground for dengue transmission [38]. The addition of temporal data regarding dengue dynamics over time will help to better understand the disease.
Within the study, we recruited 333 dengue-suspect patients from hospitals in Jakarta. The three hospitals involved are provincial public hospitals and the reference sites of primary health care centers from the Jakarta area. Approximately half (51.4%) of the samples were confirmed dengue infections, which illustrates the high burden of dengue in Jakarta. A high percentage of confirmed dengue infections during the same time period of dengue surveillance in Jakarta was also reported [33]. We notified the relatively low NS1 antigen detection sensitivity in RT-PCR-positive samples, with the lowest sensitivity was observed in DENV-4 samples. Our finding is in accordance with ours and other previous study that suggested that sensitivity of NS1 detection differed according to the serotype, and lower NS1 sensitivity was observed for DENV-4 [39, 40].
Secondary dengue infections, as inferred by serology status, accounted for most of the infections. This condition may reflect the hyperendemicity of DENV serotypes in Jakarta. In terms of the affected population, dengue infection occurred predominantly in young adults (11–20 years) (Fig. 1a). This finding is similar to the patient profile during the dengue outbreak in Jakarta in 2004 [13]. Our finding also confirms shifts in dengue infections in Indonesia from pediatric populations to young adults [11].
Examining the temporal data of DENV serotype distribution, we observed a change in the predominant serotype in Jakarta. DENV-1 was the predominant serotype circulating in the city (Fig. 1b). This is different from the serotype distribution in Jakarta in 2004, in which DENV-3 was the predominant serotype [13], and from a study from 2009 to 2010, which reported the predominance of DENV-2 [33]. The predominance of DENV-1 was also observed in other cities in Indonesia, such as in Makassar in 2007–2010 [41], Semarang in 2012 [42], and Jambi in 2015 [43]. The phenomena of serotype shifting in one city has also been reported in other cities in Indonesia [43, 44]. It has been reported that a switch in the predominant serotype was associated with outbreaks [45]. Regarding the difference in serotype predominance from other dengue study in Jakarta [33], it is possible that differences in study area, sample size, patients’ age, and serotyping method accounted for the data discordance. Compared to the other study [33], our study covered larger study area, recruited more patients including children, and used more sensitive real-time RT-PCR [19] serotyping method in addition to conventional RT-PCR. Altogether, our serotype shifting data suggested that routine active surveillance of DENV serotypes in Jakarta is needed to help build an outbreak warning system.
The determination of DENV genotypes in Jakarta was accomplished using phylogenetic analysis of the E gene of 60 closely related sequences after thorough analysis of the available sequences in the GenBank repository. The mean evolutionary rate of the DENV serotypes was within the previously reported range of 4.6–11.6 × 10−4 subs/site/year [46], with the exception of DENV-2, which exhibited a slightly higher mean evolutionary rate. However, the 95% HPD of DENV-2 still overlapped the range. The higher mean evolutionary rate in DENV-2 may reflect the high mutational rate of closely related strains within this serotype. This feature may aid the diversity of the DENV-2 Cosmopolitan genotype, which is often further grouped into different clades [31]. We also performed the Envelope gene nucleic acid similarity analyses comparing Jakarta DENV isolates with DENV strains used for dengue vaccine production [15]. We observed the mean similarities of 95.79, 93.33, 93.40, and 95.96% for DENV-1, -2, -3, and -4, respectively (data not shown). Whether this similarity will affect the vaccine efficacy warrants further studies.
The inference analysis of DENV-1 revealed the presence of Genotype I and Genotype IV. The proportion of isolates classified as Genotype I was higher than that as Genotype IV. The calculation of evolutionary parameters using Bayesian MCMC inference revealed that Genotype I of DENV-1 emerged more recently than Genotype IV, with a difference of almost a decade (Fig. 2). This result depicts the shift of the predominant genotype of DENV-1 in Jakarta, where Genotype I is actively replacing Genotype IV. This genotype shift was also reported in previous studies in other cities in Indonesia [41, 43, 44]. The predominance of Genotype I may be due to higher viral fitness, as has been previously proposed [41]. The DENV serotype/genotype shifts are of considerable potential as indicators of dengue surveillance [47]. The close relation of DENV-1 isolates with local strains and strains involved in epidemics in other countries may reflect the endemicity and diversity of DENV-1 from Jakarta and its potential spread to other regions.
The DENV-2 genotype circulating in Jakarta was the Cosmopolitan genotype, which is the common genotype in India, Southeast Asia, Africa, the Middle East, and Australia [25]. The evolutionary and relatedness analyses revealed the close relationship between Jakarta 2009 isolates and isolates from the 2004 outbreak in the same city [14] (Fig. 3). The DENV-2 isolate from concurrent infections in patients with severe dengue from Jakarta in 2013 [34] was also from the same Cosmopolitan genotype and is closely related to isolates from this study. Previous reports from other cities in Indonesia also described the grouping of DENV-2 into the Cosmopolitan genotype [30, 41,42,43, 48]. These findings indicate the endemicity of DENV-2 in Jakarta and Indonesia. We observed one isolate which was closely related to strains from Guangdong, China in 2014 (Fig. 3). It has been reported that the majority of DENV identified in China originated from Southeast Asia, mainly from Thailand, Indonesia, and the Philippines [49].
Regarding DENV-3 in Jakarta, we found that the isolates were grouped into Genotype I, which is also a common genotype found in Southeast Asia. The Jakarta DENV-3 isolates showed diverse genetic characteristics, with the grouping of individual isolates into different clades (Fig. 4). The isolates showed relatedness to isolates from Surabaya [35] and isolates from the previous 2004 and 2010 dengue epidemics in Jakarta [14, 33]. The relationship of the Jakarta isolate with strains from Singapore and imported cases in Australia and Taiwan may reflect the spread of Jakarta DENV-3. Together, these data may depict the endemicity and diversity of DENV-3 in Jakarta.
The DENV-4 Jakarta isolates were grouped into Genotype II (Fig. 5). The phylogenetic analysis revealed grouping into two clades and the close-relatedness of the isolates with strains of imported cases in Taiwan. Another study in the same time period in Jakarta [33] also observed a similar picture in which Jakarta DENV-4 isolates were grouped into the same clade. The endemicity of DENV-4 was observed with close-relatedness to strains from Indonesia, which have been circulating for more than 30 years (Fig. 5). Further surveillance is of merit to monitor the endemicity of DENV-4 in Jakarta.

Phylogenetic tree of DENV-4 isolates from Jakarta (red labels) and their most closely related reference sequences from GenBank repository. The DENV-4 Jakarta isolates were classified into Genotype II. Tree was generated by the Bayesian inference method as implemented in BEAST using the GTR + G4 + I evolution model calculated using E gene sequences. The number in the node indicates the posterior probability of that particular cluster, with values higher than 0.5 being shown (Color figure online)
We highlighted the close relationship of Jakarta isolates with strains involved in imported cases in other countries, such as Taiwan and Australia. We and others previously reported the relationship between Indonesia DENV isolates with other strains from imported cases [35, 42]. The spread of DENV between Jakarta and those countries may reflect the role of Jakarta as a major hub for trade, tourism and travel in the Southeast Asia region. Hence, it is important to monitor the endemicity of DENV in Indonesia, as it may impact epidemics in other countries [36]. Geographical expansion of dengue epidemics has become a growing threat to the health and the economy of populations living in endemic areas, where the introduction of new viral strains to regions affected by existing serotypes is a risk factor for outbreaks and severe disease [50].
We are aware of the limitations of this study, wherein only a small number of samples were assessed and samples were collected several years ago. However, our data will be beneficial for filling the gap of dengue data in Indonesia, especially in Jakarta, because the last outbreak description study with virological data was in 2004 [14]. Active DENV surveillance and monitoring in Jakarta and other cities in Indonesia should be continuous.
In summary, we have described the molecular epidemiology of dengue infection in Jakarta in 2009. We report here the change in DENV serotype predominance from DENV-3 to DENV-1 and a genotype shift in DENV-1, where Genotype I is actively replacing Genotype IV. We also report the endemicity and spread of Jakarta isolates. Our findings may add to the limited temporal data on dengue dynamics in Indonesia, as well as offer genetic information that complements current knowledge on dengue epidemiology, evolution, and transmission dynamics.
References
S. Bhatt, P.W. Gething, O.J. Brady, J.P. Messina, A.W. Farlow, C.L. Moyes, J.M. Drake, J.S. Brownstein, A.G. Hoen, O. Sankoh, M.F. Myers, D.B. George, T. Jaenisch, G.R.W. Wint, C.P. Simmons, T.W. Scott, J.J. Farrar, S.I. Hay, Nature 496, 504 (2013)
B.E.E. Martina, P. Koraka, A.D.M.E. Osterhaus, Clin. Microbiol. Rev. 22, 564 (2009)
E.A. Henchal, J.R. Putnak, Clin. Microbiol. Rev. 3, 376 (1990)
E.C. Holmes, J. Clin. Invest. 119, 2488 (2009)
E.C. Holmes, S.S. Burch, Trends Microbiol. 8, 74–77 (2000)
E.C. Holmes, S.S. Twiddy, Infect. Genet. Evol. 3, 19 (2003)
R.L. Costa, C.M. Voloch, C.G. Schrago, Infect. Genet. Evol. 12, 309 (2012)
C. Klungthong, R. Putnak, M.P. Mammen, T. Li, C. Zhang, J. Virol. Methods 154, 175 (2008)
J.P. Messina, O.J. Brady, T.W. Scott, C. Zou, D.M. Pigott, K.A. Duda, S. Bhatt, L. Katzelnick, R.E. Howes, K.E. Battle, C.P. Simmons, S.I. Hay, Trends Microbiol. 22, 138 (2014)
T.E. Setiati, J.F. Wagenaar, M.D. de Kruif, A.T. Mairuhu, E.C. van Gorp, A. Soemantri, Bull WHO 30, 1 (2006)
M.R. Karyanti, C.S.P.M. Uiterwaal, R. Kusriastuti, S.R. Hadinegoro, M.M. Rovers, H. Heesterbeek, A.W. Hoes, P. Bruijning-Verhagen, BMC Infect. Dis. 14, 412 (2014)
A.L. Corwin, R.P. Larasati, M.J. Bangs, S. Wuryadi, S. Arjoso, N. Sukri, E. Listyaningsih, S. Hartati, R. Namursa, Z. Anwar, S. Chandra, B. Loho, H. Ahmad, J.R. Campbell, K.R. Porter, Trans. R. Soc. Trop. Med. Hyg. 95, 257 (2001)
A. Suwandono, H. Kosasih, R. Kusriastuti, S. Harun, C. Ma’roef, S. Wuryadi, B. Herianto, D. Yuwono, K.R. Porter, C.G. Beckett, P.J. Blair, J. Trans. R. Soc. Trop. Med. Hyg. 100, 855 (2006)
S.H. Ong, J.T. Yip, Y.L. Chen, W. Liu, S. Harun, E. Lystiyaningsih, B. Heriyanto, C.G. Beckett, W.P. Mitchell, M.L. Hibberd, A. Suwandono, S.G. Vasudevan, M.J. Schreiber, Infect. Genet. Evol. 8, 191 (2008)
S.R. Hadinegoro, J.L. Arredondo-García, M.R. Capeding, C. Deseda, T. Chotpitayasunondh, R. Dietze, H.I.H. Muhammad Ismail, H. Reynales, K. Limkittikul, D.M. Rivera-Medina, H.N. Tran, A. Bouckenooghe, D. Chansinghakul, M. Cortés, K. Fanouillere, R. Forrat, C. Frago, S. Gailhardou, N. Jackson, F. Noriega, E. Plennevaux, T.A. Wartel, B. Zambrano, M. Saville, CYD-TDV Dengue Vaccine Working Group, N. Engl. J. Med. 373, 1195 (2015)
J.A. Usme-Ciro, J.A. Méndez, K.D. Laiton, A. Páez, Hum. Vaccin. Immunother. 10, 2674 (2014)
R.S. Lanciotti, C.H. Calisher, D.J. Gubler, G.J. Chang, A.V. Vorndam, J. Clin. Microbiol. 30, 545 (1992)
E. Harris, T.G. Roberts, L. Smith, J. Selle, L.D. Kramer, S. Valle, E. Sandoval, A. Balmaseda, J. Clin. Microbiol. 36, 2634 (1998)
R.T. Sasmono, A. Aryati, P. Wardhani, B. Yohan, H. Trimarsanto, S. Fahri, T.Y. Setianingsih, F. Meutiawati, PLoS ONE 9, e103815 (2014)
K. Katoh, G. Asimenos, H. Toh, Methods Mol. Biol. 537, 39 (2009)
A.M. Waterhouse, J.B. Procter, D.M.A. Martin, M. Clamp, G.J. Barton, Bioinformatics 25, 1189 (2009)
D.P. Martin, P. Lemey, M. Lott, V. Moulton, D. Posada, P. Lefeuvre, Bioinformatics 26, 2462 (2010)
D. Darriba, G.L. Taboada, R. Doallo, D. Posada, Nat. Methods 9, 772 (2012)
A.P. Goncalvez, A.A. Escalante, F.H. Pujol, J.E. Ludert, D. Tovar, R.A. Salas, F. Liprandi, Virology 303, 110 (2002)
S.S. Twiddy, J.J. Farrar, N. Vinh Chau, B. Wills, E.A. Gould, T. Gritsun, G. Lloyd, E.C. Holmes, Virology 298, 63 (2002)
R.S. Lanciotti, J.G. Lewis, D.J. Gubler, D.W. Trent, J. Gen. Virol. 75(Pt 1), 65 (1994)
R.S. Lanciotti, D.J. Gubler, D.W. Trent, J. Gen. Virol. 78(Pt 9), 2279 (1997)
J.-H. Huang, C.-L. Su, C.-F. Yang, T.-L. Liao, T.-C. Hsu, S.-F. Chang, C.-C. Lin, P.-Y. Shu, Am. J. Trop. Med. Hyg. 87, 349 (2012)
T. Ernst, S. McCarthy, G. Chidlow, D. Luang-Suarkia, E.C. Holmes, D.W. Smith, A. Imrie, PLoS Negl. Trop. Dis. 9, e0003442 (2015)
R. Nusa, H. Prasetyowati, F. Meutiawati, B. Yohan, H. Trimarsanto, T.Y. Setianingsih, R.T. Sasmono, J Infect. Dev. Ctries. 8, 733 (2014)
K.-S. Lee, S. Lo, S.S.-Y. Tan, R. Chua, L.-K. Tan, H. Xu, L.-C. Ng, Infect. Genet. Evol. 12, 77 (2012)
S. Churrotin, T. Kotaki, T. H. Sucipto, N. L. F. Ahwanah, P. T. Deka, K. C. Mulyatno, D. A. P. Utami, R. Ranasasmita, S. Soegijanto, and M. Kameoka, Jpn. J. Infect. Dis. 69(5), 442 (2016)
B.E. Dewi, L. Naiggolan, D.H. Putri, N. Rachmayanti, S. Albar, N.T. Indriastuti, W. Sjamsuridzal, T.M. Sudiro, Southeast Asian J. Trop. Med. Public Health 45, 53 (2014)
S. Lardo, Y. Utami, B. Yohan, S.M. Tarigan, W.D. Santoso, L. Nainggolan, R.T. Sasmono, Asian Pac. J. Trop. Med. 9, 134 (2016)
T. Kotaki, A. Yamanaka, K.C. Mulyatno, A. Labiqah, T.H. Sucipto, S. Churrotin, S. Soegijanto, E. Konishi, M. Kameoka, Jpn. J. Infect. Dis. 67, 227 (2014)
D. Warrilow, J.A. Northill, A.T. Pyke, Emerging. Infect. Dis. 18, 1850 (2012)
P.-Y. Shu, C.-L. Su, T.-L. Liao, C.-F. Yang, S.-F. Chang, C.-C. Lin, M.-C. Chang, H.-C. Hu, J.-H. Huang, Am. J. Trop. Med. Hyg. 80, 1039 (2009)
D.J. Gubler, Trop. Med. Health 39, 3 (2011)
H. Aryati, B. Trimarsanto, P. Yohan, S. Wardhani, Fahri, and R. T. Sasmono. BMC Infect. Dis. 13, 611 (2013)
M.G. Guzman, T. Jaenisch, R. Gaczkowski, V.T. Ty Hang, S.D. Sekaran, A. Kroeger, S. Vazquez, D. Ruiz, E. Martinez, J.C. Mercado, A. Balmaseda, E. Harris, E. Dimano, P.S.A. Leano, S. Yoksan, E. Villegas, H. Benduzu, I. Villalobos, J. Farrar, C.P. Simmons, PLoS Negl. Trop. Dis. 4, e811 (2010)
R.T. Sasmono, I. Wahid, H. Trimarsanto, B. Yohan, S. Wahyuni, M. Hertanto, I. Yusuf, H. Mubin, I.J. Ganda, R. Latief, P.J. Bifani, P.-Y. Shi, M.J. Schreiber, Infect. Genet. Evol. 32, 165 (2015)
S. Fahri, B. Yohan, H. Trimarsanto, S. Sayono, S. Hadisaputro, E. Dharmana, D. Syafruddin, R.T. Sasmono, PLoS Negl. Trop. Dis. 7, e2354 (2013)
S. Haryanto, R.F. Hayati, B. Yohan, L. Sijabat, I.F. Sihite, S. Fahri, F. Meutiawati, J.A.N. Halim, S.N. Halim, A. Soebandrio, R.T. Sasmono, Pathog. Glob. Health 110, 119–129 (2016)
A. Yamanaka, K.C. Mulyatno, H. Susilowati, E. Hendrianto, A.P. Ginting, D.D. Sary, F.A. Rantam, S. Soegijanto, E. Konishi, PLoS ONE 6, e27322 (2011)
K.S. Lee, Y.L. Lai, S. Lo, T. Barkham, P. Aw, P.L. Ooi, J.C. Tai, M. Hibberd, P. Johansson, S.P. Khoo, L.C. Ng, Emerg. Infect. Dis. 16, 847 (2010)
R. Chen, N. Vasilakis, Viruses 3, 1562 (2011)
S. Runge-Ranzinger, P.J. McCall, A. Kroeger, O. Horstick, Trop. Med. Int. Health 19, 1116 (2014)
T. Kotaki, A. Yamanaka, K.C. Mulyatno, S. Churrotin, T.H. Sucipto, A. Labiqah, N.L.F. Ahwanah, S. Soegijanto, M. Kameoka, E. Konishi, Infect. Genet. Evol. 37, 88 (2016)
S. Sang, B. Chen, H. Wu, Z. Yang, B. Di, L. Wang, X. Tao, X. Liu, Q. Liu, Infect. Genet. Evol. 32, 178 (2015)
A. Kroeger, M. Nathan, J. Hombach, Nat. Rev. Microbiol. 2, 360 (2004)
Acknowledgements
The authors are grateful to all the patients and clinicians involved in this study and the help of colleagues from the immunology and virology laboratories of the Center for Research and Development of Biomedical and Basic Health Technology, NIHRD, Indonesia Ministry of Health. This study was funded by the NIHRD, Indonesia Ministry of Health, and a SINAS grant from the Indonesia Ministry of Research, Technology, and Higher Education.
Author information
Authors and Affiliations
Contributions
CSWL and RTS conceived and designed the study. CSWL and AY collected the samples. BY, AY, FM, and RFH performed the experiments. CSWL, BY, HT, and RTS analyzed the data. BY wrote the first draft. CSWL, HT, and RTS edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
All authors declare that they have no conflict of interest.
Research involving human participants and/or animals
Involvement of human subjects in this study was approved by the National Institute for Health Research and Development (NIHRD) Research Ethics Commission, ethics approval number LB.03.02/KE/4537/2009—June 24, 2009.
Informed consent
Written informed consents were obtained from the patients or their legal guardians.
Additional information
Edited by Lorena Passarelli.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lestari, C.S.W., Yohan, B., Yunita, A. et al. Phylogenetic and evolutionary analyses of dengue viruses isolated in Jakarta, Indonesia. Virus Genes 53, 778–788 (2017). https://doi.org/10.1007/s11262-017-1474-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-017-1474-7