
Abstract
Grapevine Syrah virus-1 (GSyV-1) was identified by small-RNA deep sequencing in Slovak grapevine co-infected by several other viruses. The RT-PCR assays developed in this work substantially improved the virus detection and allowed the identification of GSyV-1 in tested grapevine samples from Slovakia and the Czech Republic at an unexpectedly high rate (ca. 30 %). Subsequently, complete genome sequences of 3 GSyV-1 isolates (2 Slovak and 1 Czech) were determined by Sanger sequencing, showing a typical marafivirus genome organization. Analyses of complete genome sequences showed a higher intra-group diversity among these 3 central European GSyV-1 isolates (differences reaching 7.1 % at the nucleotide level) in comparison to 3 previously characterized North American isolates (only 1.2 % intra-group divergence). A substantially higher divergence among central European isolates and their clustering into two major phylogenetic groups was further confirmed by the partial genome analysis of additional 26 isolates. The CP-centered study did not support the geography-based clustering among central European and American isolates. Nevertheless, the sequence data of the highly variable 5′-proximal portion of the genome obtained for few additional isolates from Slovakia and Czech Republic showed the presence of both, “European-” and “north American-like”, GSyV-1 isolates in the analyzed grapevine samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
More than 70 graft-transmissible infectious agents have been recorded from grapevine, many of which are associated with economically important diseases [1]. Nevertheless, this number is destined to grow in the future due to two main factors: (i) increased interest in grapevine virus research in many laboratories worldwide, (ii) recent development of powerful, unbiased and high throughput next-generation sequencing (NGS; synonym “Deep sequencing”) technologies.
NGS technologies, developed at the onset of 21st century, have revolutionized field of genomics and have contributed to ground-breaking discoveries in many branches of science (i.e., medicine, ecology, marine biology), including virology. “Deep sequencing” coupled with metagenomic approaches opened unimaginable opportunities for the in extenso studies concerning virus biodiversity and ecology in natural [2, 3] and/or agricultural ecosystems [4], as well as for discovery and characterization of new viruses associated with certain diseases [5, 6]. Several NGS-based studies have resulted in the identification of several novel viruses of cultivated crops, including grapevine [1, 7].
NGS analysis of a cv. Syrah grapevine from California (USA), affected by severe decline syndrome, revealed a mixed infection of several viruses and viroids, including an unknown marafivirus, named Grapevine Syrah virus-1 [8]. Nevertheless, low level (19 %) of GSyV-1 association with the disease [8] indicated unlikely involvement of this virus in the Syrah Decline problem.
In an independent and contemporary study of viruses affecting native grapes in southern USA, the same virus was identified in a muscadine grape (Vitis rotundifolia Michx.) from Mississippi and reported under the name Grapevine virus Q [9]. The same study reported identification of an isolate of GVQ in wild blackberries in Great Smoky Mountains, indicating that virus host range is not restricted only to Vitis spp. Detailed analyses of GVQ genome reported in that study revealed characteristic “permuted” organization of RdRp motifs, a feature unreported in other plant viruses [9].
The two complete sequences determined for GSyV-1 and GVQ (FJ977041, FJ436028) shared mutually more than 99 % nucleotide identity unambiguously confirming that the two viruses belong to the same, novel species in the genus Marafivirus in the family Tymoviridae, for which the name GSyV-1 was officially adopted by the International Committee on Taxonomy of Viruses [10].
The presence of GSyV-1 was subsequently reported from Chile [11] and from Washington State in the USA [12]. However, the knowledge on GSyV-1 outside the American continent is still very scarce and limited to few published papers. Giampetruzzi et al. [13] identified the GSyV-1 in Italy while analysing NGS dataset generated from a grapevine simultaneously infected by several viruses. In a separate study, RT-PCR based survey frequently identified this virus in a clonal material of cv. Syrah in France [14]. All of the afore-mentioned studies from outside of the USA report only partial nucleotide sequence data for studied GSyV-1 isolates (250 and 750 nucleotides). However, the search in the GenBank (January 2015), revealed the availability of complete nucleotide sequences of another GSyV-1 isolate from Canada (isolate 3138-01, GenBank Acc. No. JX13896.1).
In the present work, GSyV-1 was originally detected in a grapevine accession SK30 from Slovakia using NGS approach and, subsequently, an improved RT-PCR protocol was developed and applied to investigate its incidence in grapevine samples from Slovakia and Czech Republic. Finally, three GSyV-1 isolates from grapevines collected in Slovakia and Czech Republic have been completely sequenced and partial genome sequence data for several additional isolates were determined, allowing to assess the GSyV-1 genetic diversity.
Materials and methods
Plant samples
The samples analyzed in the present study were collected during the period 2010–2014, from grapevine plants grown in different locations in Slovakia or they originated from the collection located at Crop Research Institute, Ruzyne, Prague, Czech Republic. Total RNAs were extracted either from cortical scrapings from dormant canes or leaf petioles collected during the vegetation period, using the Nucleo Spin RNA Plant kit (Macherey–Nagel, Düren, Germany) following the manufacturer’s instructions.
Next-generation sequencing
Twenty year old white-berry cv. Veltliner (referred to as SK30), previously ascertained to host several viruses [15], was subjected to siRNA next-generation sequence analyses. siRNAs were isolated from 0.1 g of fully expended leaves from different parts of the plant using the mirPremier microRNA Isolation Kit (Sigma-Aldrich, St. Louis, USA) and quantified with the Qubit® 2.0 Fluorometer (Invitrogen, Grand Island, USA) and a Bioanalyzer 2200 TapeStation (Agilent Technologies, Santa Clara, USA). Subsequently, siRNAs were sequenced by LifeSequencing S.L. (Paterna, Spain) using TrueSeq v.3 Illumina technology on a HiSEq 1000 instrument (Illumina, San Diego, USA). De novo genome assembly was performed with the Geneious (Biomatters Ltd., Auckland, New Zealand), CLC Genomics Workbench (CLC bio, Aarhus, Denmark), and Velvet (command-line) (http://www.ebi.ac.uk/~zerbino/velvet/) software. The resulting contigs were blasted (Blastn, BlastX, TBlastX) against nucleotide and protein databases at NCBI. Mapping of siRNAs against the reference genomes of viral species detected in de novo assembly was performed with the Geneious software suite with different custom mapping options.
RT-PCR-based detection
Two sets of primers were used for the study of GSyV-1 incidence in grapevine samples collected in Slovakia and Czech Republic. A pair of primers reported by Al Rwahnih et al. [8], targeting a 297-bp fragment of the putative methyltransferase gene (GSyV-1Det-F: 5′-CAAGCCATCCGTGCATCTGG-3′, sense and GSyV-1Det-R: 5′-GCCGATTTGGAACCCGATGG-3′, antisense) was used in the initial phases of the survey.
A novel set of detection primers, SY5922F (5′-CCAATGGGTCGCACTTGTTG-3′, sense) and SY6295R (5′-ACTTCATGGTGGTGCCGGTG-3′, antisense), was designed on the available GSyV-1 sequences in the Genbank database (www.ncbi.nlm.nih.gov) and targeted a 374 bp fragment of the CP gene. This set of primers was used later in this work to validate initial survey data.
For both primer combinations, a two-step RT-PCR protocol was used. The first-strand cDNA was synthesized using random hexamer primers and the PrimeScript (MMLV) reverse transcriptase (TaKaRa, Bio Inc., Shiga, Japan) used as recommended by the supplier. PCR amplification was performed using the proofreading TaKaRa Ex Taq polymerase (TaKaRa, Bio Inc., Shiga, Japan) under the following cycling conditions: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 20 s, 56 °C for 20 s, and 72 °C for 30 s; final extension step at 72 °C for 10 min. All PCR products were purified using the Wizard SV Gel and PCR Clean-up System (Promega Corp., Madison, WI, USA) and directly sequenced in both directions using an automated DNA sequencer (ABI 3130xl Genetic Analyser; Applied Biosystems).
Grapevine samples tested for GSyV-1 infections were also checked for the presence of 5 additional viruses via RT-PCR using specific primers. The viruses tested for and primer sequences are: Arabis mosaic virus, ArMV (Ar2693F 5′-AGTGCYTACAAGAGTGTGTC-3′, sense and Ar3746R 5′-CAACACACTGTCGCCACT-3′ antisense, Glasa unpublished), Grapevine fleck virus, GFkV [16], Grapevine leafroll-associated virus-1, GLRaV-1 [17], Grapevine Pinot gris virus, GPGV [15] and Grapevine virus A, GVA (GVA6535F 5′-CTCTCTTYGGGTACATCGC-3′, sense and GVA7180R 5′-CTTCCCTCTTTCACGAACC-3′antisense, Glasa unpublished).
Determination of the full-length genome sequence of GSyV-1 isolates using Sanger sequencing
Complete genomic sequences of the SK30, SK413, and CZ10 isolates were obtained by spanning the genome with several overlapping PCR fragments amplified using the TaKaRa LA Taq polymerase (TaKaRa, Bio Inc., Shiga, Japan) and the custom-made primers designed on the previously available complete genome sequences (FJ977041, FJ436028, JX513896) (all primer sequences are available from the author). All PCR products were directly sequenced. When necessary, the genome portions were verified by an additional PCR amplifications and sequencing employing different set of primers (Supplementary Table 1). The 5′-end of the genome was amplified using a 5′-Full RACE Core Set kit (TaKaRa, Bio Inc., Shiga, Japan) with the 5′-end phosphorylated antisense primer SY-692P (5′-CGTGCTGCAACTTC-3′) and four gene-specific primers SY-172S2 (5′-GGAAGCGTAAGCGAAAGC-3′), SY-256S1 (5′-ATAGCGGAGGCTATGAGC-3′), SY-381A1 (5′-GTCTCAAGGAATTGGTCG-3′), and SY-584A2 (5′-CACAAGACGATCGAGATC-3′), following the manufacturer’s instructions.
Sequence analyses
Partial and complete sequences were compared with the sequences available in the GenBank database (www.ncbi.nlm.nih.gov). Sequence analyses were performed using either MEGA v.6 [18] or on-line tools (http://web.expasy.org/compute_pi/, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) [19]. The phylogenetic trees were inferred using the neighbor-joining algorithm implemented in MEGA v.5 and strict identity distance metrics for both nucleotide and amino acid sequence analyses.
Results
NGS data analyses and identification of GSyV-1
The analyses of siRNA pool and de novo assembly of contigs revealed a mixed infection of SK30 grapevine with several known viruses: ArMV, GFLV, GDefV, GRSPaV, GFkV, GLRaV-1, and GLRaV-3 [15 and unpublished results]. Interestingly, analysis of siRNA reads revealed the presence of grapevine Pinot gris virus (GPGV) and GSyV-1, both previously undetected in Slovakia or in any other country in Central Europe. As characterization of GPGV was subject of a previous paper [15] here, we focus on characterization of GSyV-1.
A total of 3469,390 high-quality reads, ranging from 21–24 nucleotides in size, were used for the bioinformatics analysis. De novo assembly resulted in a total of 552 contigs between 47 and 674 nucleotides in size. Sixty-five percent of contigs (356 out of 552) were identified as viral (55,102 siRNAs), of which 1 contig was specific to GSyV-1. Mapping of individual siRNAs against reference GSyV-1 sequence (FJ436028) enabled the partial reconstitution of GSyV-1 full-length sequence, covering 5113 nucleotides or 78.8 % of the genome (2182 siRNAs with a mean size of 21.2 nucleotides).
Study of GSyV-1 incidence in grapevine samples in Slovakia and Czech Republic
The discovery of GSyV-1-like contig in the analyzed NGS dataset prompted further study to evaluate the occurrence and incidence of GSyV-1 in the central European region by testing a total of 95 samples from Slovakia and Czech Republic. A primer pair GSyV-1Det-F/GSyV-1Det-R [8], used in the initial phase of the survey, detected this virus in 15 out of 95 tested samples (12 of 85 grapevine samples from various localities in Slovakia and in 3 of 10 samples from Czech Republic). The sequence analysis of Slovak and Czech isolates showed nucleotide identities ranging from 93.4 to 96.9 % with reference FJ436028 isolate in this short genomic region (for acc. numbers see Table 1).
In order to verify and possibly ameliorate the accuracy of GSyV-1 identification in the absence of wide spectrum commercial serological tests, a new pair of primers SY1036F/SY1832R targeting different regions of the GSyV-1 genome (i.e., the CP gene) was designed and tested in this work. Interestingly, the re-testing of the same sample set initially analyzed by GSyV-1Det-F/GSyV-1Det-R resulted in a substantially higher number of GSyV-1 infected samples (28 vs. 15, see Table 1). Therefore, the primer set targeting viral CP confirmed the presence of GSyV-1 in all 15 grapevines resulted positive with MTR-based set and revealed infections of this virus in additional 13 samples.
To eliminate the possibility of any experimental bias, whole sample set was again reverse transcribed and repeatedly re-tested using both primer sets. However, the new experiments confirmed the previous observation indicating much better suitability of the new primer set for GSyV-1 detection in our conditions. Two isolates, SK410 and SK411, not recognized by GSyV-1Det-F/GSyV-1Det-R were therefore amplified using new primer set SY1036F/SY1832R overlapping the GSyV-1Det-F/GSyV-1Det-R portion and sequenced (acc. numbers KP236107 and KP236108). As expected, the examination of nucleotide sequence data showed several mismatches in the 5′ terminal sequence of both primers (GSyV-1Det-F/GSyV-1Det-R) which was likely the reason of the failure to detect the virus with this primer set.
Overall, 25 of 85 samples from Slovakia and 3 of 10 samples from Czech Republic were tested positive for GSyV-1 using this newly designed primer set (Table 1).
Molecular diversity among GSyV-1 isolates in the CP gene
The 29 partial CP sequences of GSyV-1 generated in this work (SK30 plus 28 samples identified in the survey) of 334 bp in size after primer removal, were used to assess the genetic variability of Slovak and Czech isolates and to study their relationship with the previously characterized isolates from other parts of the world. Multiple alignment showed that all GSyV-1 sequences were fully collinear and without indels.
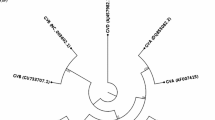
Phylogenetic analysis revealed that GSyV-1 isolates grouped in two major clades. Most of GSyV-1 isolates used in this study (37 out of 40) grouped together to form a major clade composed of two major lineages (Fig. 1). The three isolates from Central Europe (SK412, SK415, and CZ10) were found divergent in this part of genome and formed a separate cluster. Similar patterns of sequence relationships were obtained when other algorithms (maximum parsimony, minimum evolution) were used for phylogenetic reconstruction (data not shown).

The neighbor-joining phylogenetic tree generated from the partial GSyV-1 CP gene sequences using strict identity distance metrics. The previously characterized isolates are identified by their accession numbers and geographical origin. Isolates sequenced in this study are letter-coded as SK or CZ. Bootstrap values >70 % (1000 bootstrap resamplings) are indicated on the branches. Grapevine rupestris vein feathering virus (GRVFV, GenBank accession number AY706994) was used to root the tree
The analysis of different geographical groups based on the partial CP sequences revealed that the within-group mean genetic diversity (p-distance model) of 6.0 % (±0.8 %) for Slovak isolates (25 sequences available) and 8.0 % (±1.1 %) for Czech isolates (3 sequences). The divergence calculated within 15 GSyV-1 isolates from Washington State (USA) was lower and reached 3.9 % (±0.6 %).
Analysis of the complete genome sequences of three new GSyV-1 isolates from Central Europe
The genomes of the three GSyV-1 isolates consist of 6481 nucleotides (SK413) or 6482 nucleotides (SK30, CZ10), excluding the polyA tail. Similarly to the reference sequence FJ436028, the three studied genomes contain an ORF (6243 nucleotides) and non-coding regions of 141/142 and 97 nucleotides at their 5′ and 3′ termini, respectively. The marafibox sequence [20], reported to be transcription promoter for the synthesis of subgenomic RNA, was found in all sequences, starting at the expected nucleotide position 5698 (SK413) or 5699 (SK30, CZ10).
The amino acid sequence of the polyproteins deduced form the large ORF of all isolates are strictly collinear (2081 amino acids in length), and contain the motifs similar to those previously identified in marafiviruses, i.e., metyltransferase, proteinase, helicase, and RNA-dependent RNA polymerase [10]. The C-terminal region of the polyprotein includes two co-terminal capsid proteins of ca. 23 and 21 kDa, signature of marafiviruses (Fig. 2a).

a Schematic representation of the GSyV-1 genome with major nucleotide coordinates. GSyV-1 is typified by the isolate SK30. Boxes represent the two ORFs identified, whereas the lines represent untranslated regions. Conserved amino acid domains in the ORF1 are indicated. MTR viral methyl transferase, PRO protease, Hel helicase, RdRp RNA-dependent RNA polymerase, CP coat protein. b Nucleotide diversity index between the aligned reference GSyV-1 sequence (FJ436028) and SK30, SK413, and CZ10 genomic sequences, calculated along the genome using DnaSP software with a sliding window of 100 nucleotides moved by steps of 25 nucleotides
The deduced molecular masses of 2081 amino acid long polyproteins of three studied isolates were 229.678 (SK30), 229.511 (SK413), and 229.700 kDa (CZ10).
The Slovak and Czech isolates share 91.9–93.3 % identity at the nucleotide level with the previously characterized ones (Table 2). In the phylogenetic analysis they are clearly separated from the north American isolates which form a coherent and highly supported clade (Fig. 3). Interestingly, while the nucleotide divergence among three complete genomes of north American isolates is only 1.2 % (±0.1 %), the divergence among 3 Slovak and Czech sequences is substantially higher and reached 7.1 % (±0.2 %) (see Supplementary Table 2 for details).

Phylogenetic tree generated on complete nucleotide genome sequences of marafiviruses: Grapevine rupestris vein feathering virus (GRVFV), Grapevine Syrah virus-1 (GSyV-1), Blackberry virus S (BlVS), Oat blue dwarf virus (OBDV), Citrus sudden death-associated virus (CSDaV), Olive latent virus-3 (OLV-3), Maize rayado fino virus (MRFV), and Switchgrass mosaic virus (SwMV). GenBank Accession numbers for all viruses/isolates used in the analysis are indicated. The three Slovak and Czech GSyV-1 isolates sequenced in the present study are highlighted in bold. The scale bar indicates a genetic distance of 0.1. Bootstrap values (1000 bootstrap re-samplings) are indicated on the branches as percentages. The phylogenetic tree was reconstructed using the neighbor-joining algorithm implemented in MEGA v.6. Grapevine fleck virus (GFkV, gen. Maculavirus) was used as an outgroup
The comparison of determined sequences of two Slovak and a Czech isolate with a reference FJ436028 sequence using a sliding window analysis of nucleotide diversity revealed that the variability among the isolates was not evenly distributed along the entire genome, but rather concentrated in few “hot spots” with divergence reaching more than >20 % (Fig. 2b). The most divergent genome regions were the 5′ UTR (nt 94–135) and the beginning of ORF (nucleotides 273–374, subsequently affecting the amino acids 42–73), where the identities of SK413, CZ10, and SK30 with the reference FJ436028 at the nucleotide level reached only 50.0, 53.9, and 61.8 % (at amino acid level 30.0, 33.3, and 36.7 %, respectively). The blastn and blastp searches, however, did not revealed any similarities with a known sequence that would be indicative of a potential inter-species recombination event in this polyprotein portion.
The nucleotide sequence data of the highly variable 5′ extremity of the genome (nucleotides 37–654) were determined for additional 3 isolates. Interestingly, while the SK104 isolate is phylogenetically related to other sequenced Slovak and Czech isolates (Supplementary Fig. A), the SK416 and CZ01 isolates were more similar to the north American isolates. SK416 and CZ01 isolates shared 96.6–96.8 % nucleotide identities with American isolates while being only 78.6–82.0 % identical to the corresponding regions of completely sequenced SK30, SK413, and CZ10 isolates.
As previously reported for this virus [9], three studied GSyV-1 isolates had a characteristic permuted C → A → B sequence motifs forming the active site of putative RNA-dependent RNA polymerase at identical amino acid positions (C: 1585–1597, A: 1604–1614, B: 1650–1677) as in the north American GSyV-1 isolates (FJ977041, FJ436028, JX513896). The deduced amino acid sequences of the C → A → B motifs were identical for all isolates.
An additional putative ORF of 804 nucleotides present in three US sequences (FJ436028, FJ977041, JX513896) was also found in the SK30, SK413, and CZ10 genomes. While in SK30 sequence, the ORF starts at the same position as previously reported (nucleotide position 752), this ORF starts at nucleotide position 695 in SK413 and CZ10, thus the ORF is 57 nucleotides longer. The identities with reference FJ436028 isolate were 75.7, 83.5, and 88.8 % for SK30, SK413, and CZ10. Interestingly, while the amino acid divergence among 3 US isolates in the deduced amino acid sequence is only 1.2 % (±0.5 %), the divergence among SK and Czech isolates reached 21.4 % (±2.0 %). The biological significance of the variation in this putative protein as well as its possible function (if any) is yet unknown.
Discussion
Novel DNA sequencing techniques, referred to as “next-generation” sequencing, provide high speed and high throughput, generating an enormous volume of sequence data with numerous applications, such as detection and identification of known viruses and/or viroids in infected plants or powerful characterization of novel pathogen(s) without an a priori knowledge [5, 21].
siRNA-based deep sequencing approach has previously been used to characterize viral genomes in several host plants [6, 22], including grapevine [13, 15, 23, 24]. Similar approach (illumina sequencing of small, virus-related RNA molecules), applied in this work, allowed initial identification of a large contig related to Grapevine Syrah virus-1 in a grapevine sample SK30 from Slovakia. This finding represents an original report of this virus in Central Europe that triggered further study focused on GSyV-1 in this part of the world.
However, the genome of a Slovak GSyV-1 isolate could be reconstructed only partially from the original deep-sequenced siRNAs by mapping against reference isolate. It suggests that the number of reads of 21–24 nucleotides was insufficient to fully cover GSyV-1 genome, due to its low initial concentrations in a studied grapevine sample affected with mixed viral infection.
Using standard dideoxy sequencing approach, we completely sequenced genomes of three new GSyV-1 isolates (2 from Slovak grapevines and one from Czech Republic). This work represents the first report of complete nucleotide sequences of GSyV-1 originating from the European continent and furthers the overall knowledge on this virus on a global scale.
Results of the analyses suggest that GSyV-1 population from Central Europe is genetically distinct from the American isolates. The three fully sequenced American isolates of GSyV-1 appear virtually identical from the molecular standpoint despite the fact that they originated from very distant and distinct geographic locations (California and Mississippi in USA and British Columbia in Canada), as well as from diverse hosts (muscadine and grapevine) [8, 9]. The most diverged American isolates (from Canada and Mississippi) differed less than 2 % at the nucleotide sequence level and only 1.3 % in amino acids content. On the other side, all three isolates from Central Europe sequenced in this work appeared distinct as they differed 6–8 % in nucleotide content when compared from the “American group”. Furthermore, the similar level of diversity was found among the three studied isolates from Slovakia and Czech Republic, suggesting that GSyV-1 population in Central Europe is genetically more diverse than the North American one.
Variability of central European GSyV-1 population compared to the North American isolates was further confirmed on the larger dataset. The analysis of partial CP sequences of 28 GSyV-1 isolates from two Central European countries revealed that the within-group mean genetic diversity (p-distance model) reached 6.0 % (±0.8 %) for Slovak isolates (25 sequences available) and 8.0 % (±1.1 %) for Czech isolates (3 sequences). The divergence calculated within 15 GSyV-1 isolates from Washington State USA [12] was lower and reached 3.9 % (±0.6 %), indicative of a more genetically homogenous viral population.
The determination of partial sequences of the 5′extremity of the genome (nucleotides 37–654) from three additional isolates (SK104, SK416 and CZ01) suggests the presence of the “American-like” GSyV-1 genotypes in both in Slovakia and Czech Republic along with variants similar to SK30, SK413 and CZ10 (Supplementary Fig. A and unpublished data). Curiously, the distinction of American and central European isolates observed in the analyses generated from the complete (Fig. 3) or the 5′-proximal nucleotide sequences (Supplementary Fig A), is not reflected in the phylogenetic tree generated from the partial CP sequences (Fig. 1) being more conserved among isolates. This confirms that the focus only on short genomic region might not be suitable for accurate genotyping of virus isolates due to insufficient number of informative sites and possible intra-species recombinations.
This study revealed relatively high incidence (29.5 %) of GSyV-1 in a range of tested white- and red-berried grapevine cultivars from Slovakia and Czech Republic. However, no particular symptom could be attributed to the GSyV-1, as most of the grapevine plants analyzed in this work were simultaneously infected by other viruses, as determined by specific RT-PCRs targeting ArMV, GFkV, GLRaV-1, GPGV, GVA (Table 1). This is not unusual, as co-infections of few viruses were often found in single grapevines in surveys carried out in several countries. Nevertheless, the unbiased and high throughput NGS-based studies have further highlighted complexity of plant viromes, especially in case of perennial host such as grapevine [4, 8, 13, 15, 24–26]. This complicates the establishment of links between the symptoms observed and the presence of a given infectious agent. Therefore, the real impact, if any, of GSyV-1 on the Slovak and Czech grapevines remains yet to be studied.
Finally, our study highlighted the importance of reliable and robust diagnostic protocol for routine detection of viruses. Primer set SY5922F/SY6295R, designed in this study, allowed GSyV-1 detection in nearly double number of positive samples (28 vs. 15) compared to primer pair GSyV-1Det-F and GSyV-1Det-R reported in bibliography [8] and initially used for diagnosis of the virus. The dramatic differences in primer performance could be due to poor conservation of the genome portion targeted by primers reported by Al Rwahnih and collaborators [8]. Indeed, the initial 1000 nucleotides represent the most variable genomic portion in many viruses, including GSyV-1 (see Fig. 2b). Indeed, our analyses revealed several nucleotide mismatches present in the GSyV-1 Det-F/Det-R target region in genomes of Slovak isolates which resulted in failure of their detection by this set of primers. On the other side, primers SY5922F/SY6295R were designed on highly conserved regions of the viral CP gene in several isolates. An additional advantage of newly designed primers is that they have an “enriched” template pool in GSyV-1 infected plants, represented by full genomic size molecules as well as by CP-specific subgenomic RNAs known to be produced during infections by marafiviruses [10]. Thus, the primer set SY5922F/SY6295R may enable a more effective routine detection and monitoring of GSyV-1 in vineyards and nurseries.
References
G.P. Martelli, J. Plant Pathol. 96, 1–136 (2014)
M.J. Roossinck, P. Saha, G.B. Wiley, J. Quan, J.D. White, H. Lai, F. Chavarria, G. Shen, B.A. Roe, Mol. Ecol. 19(Suppl 1), 81–88 (2010)
M.J. Roossinck, Annu. Rev. Genet. 46, 359–369 (2012)
B. Coetzee, M.-J. Freeborough, H.J. Maree, J.-M. Celton, D.J.G. Rees, J.T. Burger, Virology 400, 157–163 (2010)
I.P. Adams, R.H. Glover, W.A. Monger, R. Mumford, E. Jackeviciene, M. Navalinkiene, M. Samuitiene, N. Boonham, Mol. Plant Pathol. 10, 537–545 (2009)
J.F. Kreuze, A. Perez, M. Untiveros, D. Quispe, S. Fuentes, I. Barker, R. Simon, Virology 388, 1–7 (2009)
M. Barba, H. Czosnek, A. Hadidi, Viruses 6, 106–136 (2014)
M. Rwahnih, S. Daubert, D. Golino, A. Rowhani, Virology 387, 395–401 (2009)
S. Sabanadzovic, N. Abou Ghanem-Sabanadzovic, A.E. Gorbalenya, Virology 394, 1–7 (2009)
T.W. Dreher, M.C. Edwards, A.-L. Haenni, R.W. Hammond, I. Jupin, R. Koenig, S. Sabanadzovic, G.P. Martelli, Family Tymoviridae, in Virus Taxonomy (IXth Report of the ICTV), ed. by A.M.Q. King, E. Lefkowitz, M.J. Adams, E.B. Carstens (Elsevier Academic Press, San Diego, 2012), pp. 913–921
E.A. Engel, P.A. Rivera, P.D.T. Valenzuela, Plant Dis. 94, 633 (2010)
T.A. Mekuria, R.A. Naidu, Plant Dis. 94, 787 (2010)
A. Giampetruzzi, V. Roumi, R. Roberto, U. Malossini, N. Yoshikawa, P. La Notte, F. Terlizzi, R. Credi, P. Saldarelli, Virus Res. 163, 262–268 (2012)
M. Beuve, B. Moury, A.-S. Spilmont, L. Sempé-Ignatovic, C. Hemmer, O. Lemaire, Eur. J. Plant Pathol. 135, 439–452 (2013)
M. Glasa, L. Predajňa, P. Komínek, A. Nagyová, T. Candresse, A. Olmos, Arch. Virol. 159, 2103–2107 (2014)
M. Glasa, L. Predajňa, P. Komínek, J. Phytopathol. 159, 805–807 (2011)
L. Predajňa, A. Gažiová, E. Holovičová, M. Glasa, Acta Virol. 57, 353–356 (2013)
K. Tamura, G. Stecher, D. Peterson, A. Filipski, S. Kumar, Mol. Biol. Evol. 30, 2725–2729 (2013)
A. Marchler-Bauer, S. Lu, J.B. Anderson, F. Chitsaz, M.K. Derbyshire, C. DeWeese-Scott, J.H. Fong, L.Y. Geer, R.C. Geer, N.R. Gonzales, M. Gwadz, D.I. Hurwitz, J.D. Jackson, Z. Ke, C.J. Lanczycki, F. Lu, G.H. Marchler, M. Mullokandov, M.V. Omelchenko, C.L. Robertson, J.S. Song, N. Thanki, R.A. Yamashita, D. Zhang, N. Zhang, C. Zheng, S.H. Bryant, Nucleic Acids Res. 39, D225–D229 (2011). doi:10.1093/nar/gkq1189
N. Abou Ghanem-Sabanadzovic, S. Sabanadzovic, G.P. Martelli, Virus Genes 27, 11–16 (2003)
S. Massart, A. Olmos, H. Jijakli, T. Candresse, Virus Res. 188, 90–96 (2014)
J. Seguin, R. Rajeswaran, N. Malpica-López, R.R. Martin, K. Kasschau, V.V. Dolja, P. Otten, L. Farinelli, M.M. Pooggin, PLoS ONE 9, e88513 (2014)
V. Pantaleo, P. Saldarelli, L. Miozzi, A. Giampetruzzi, A. Gisel, S. Moxon, T. Dalmay, G. Bisztray, J. Burgyan, Virology 408, 49–56 (2010)
Y. Zhang, K. Singh, R. Kaur, W. Qiu, Phytopathology 101, 1081–1090 (2011)
M. Al Rwahnih, S. Daubert, J.R. Urbez-Torres, F. Cordero, A. Rowhani, Arch. Virol. 156, 397–403 (2011)
S. Poojari, O.J. Alabi, V.Y. Fofanov, R.A. Naidu, PLoS ONE 8, e64194 (2013)
Acknowledgments
This work was supported by Grant VEGA 2/0060/13 from the Scientific Grant Agency of the Ministry of Education and Slovak Academy of Sciences. AO received the support from the INIA project RTA2011-00067-C04. KŠ was partially supported by the Grant REVOGENE (ITMS 26240220067) supported by the Research & Development Operational Programme funded by the ERDF. We thank Dr. P. Kominek for providing the grapevine samples from the Czech Republic.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Karel Petrzik.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11262_2015_1201_MOESM2_ESM.pptx
Supplementary Fig. A Phylogenetic tree generated from the sequences encompassing the 5′end proximal part of the genome (nucleotides 37-654). (pptx 33 kb)
Rights and permissions
About this article

Cite this article
Glasa, M., Predajňa, L., Šoltys, K. et al. Detection and molecular characterisation of Grapevine Syrah virus-1 isolates from Central Europe. Virus Genes 51, 112–121 (2015). https://doi.org/10.1007/s11262-015-1201-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-015-1201-1