
Abstract
The complete genome sequence of a novel norovirus strain GZ2010-L87 identified in Guangzhou was analyzed phylogenetically in this study. The RNA genome of the GZ2010-L87 strain is composed of 7,559 nucleotides. The phylogenetic analysis based on open reading frame (ORF) 2 revealed that the strain belongs to the GII.4 genotype, forming the new cluster GII.4-2009 which was also identified in Asia and the USA since 2009. Furthermore, phylogenetic analyses of the full genome and the different open reading frame sequences of GZ2010-L87 and other representative strains suggested that the novel strain did not undergo recombination. Comparative analysis with the consensus sequence of 31 completely sequenced norovirus GII.4-2009 genomes showed 86 mismatched nucleotides (56 in ORF1, 16 in ORF2, and 14 in ORF3), resulting in 19 amino acid changes (9 in ORF1, 3 in ORF2, and 7 in ORF3). Furthermore, 12 variable sites were found on the capsid protein of norovirus GII.4-2009, and most were located at the P2 domain. Meanwhile, based on comparison with other GII.4 clusters, 14 sites were shown specific to the novel cluster. In summary, the genome of the new GII.4-2009 variant GZ2010-L87, which was first identified in China, was extensively characterized with a large panel of genetically diverse noroviruses. The genomic information obtained from the novel variant can be used not only as a full-length norovirus sequence standard in China but also as reference data for future evolution research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Norovirus (NoV) is the most important causative agent of non-bacterial acute gastroenteritis in humans of all age groups, resulting in heavy economic impacts because of its ubiquitous nature [1, 2]. Almost 267 million NoV infections have been identified worldwide, with nearly 220,000 children dying in developing countries every year [3, 4]. NoVs could spread through person-to-person contact as well as contaminated food, water, and environment [5, 6]. In addition to its multiple modes of transmission, NoVs are highly contagious with a prolonged period of environmental persistence and low infection dosage [7, 8]. Preventing NoVs from spreading is difficult once introduced, and outbreaks are frequently reported in semi-closed or closed institutions, such as hospitals, nursing homes, schools, restaurants, ships, and military bases [9].
NoVs belong to the family Caliciviridae, with a positive sense single-stranded RNA genome, 7.5–7.7 kb in size, consisting of three open reading frames (ORFs) with a poly-A tail at the 3′-end. ORF1 encodes a polyprotein that is cleaved into six non-structural proteins [10]. ORF2 encodes a major structural protein VP1, which consists of a shell domain and a protruding arm that is further divided into two subdomains, P1 and P2 [11, 12]. ORF3 encodes a minor protein VP2, which is rich in basic amino acids and essential for viral stability and expression [13].
Based on VP1 amino acid sequences, NoVs are classified into five genogroups, among which GI, GII, and GIV have the ability to infect humans [14]. Each genogroup could be further subdivided into different genotypes, including 9 in GI and 19 in GII, of which GII.4 is the predominant strain causing majority of the outbreaks and sporadic infections worldwide [15, 16]. Furthermore, accumulated mutations in the P2 domain have generated different GII.4 NoVs variants in the past 3 decades [17, 18].
Currently, no suitable in vitro reproduction and culture system exists for human NoVs. Thus, verification of the characteristics of NoVs based on the analysis of the entire sequence will aid in the accurate analyses and evaluation of the epidemiological and molecular features as well as the discovery of new antigenic types of viral groups [19, 20]. The importance of full-length NoV genome sequencing has recently drawn more attention worldwide [21]. However, in developing countries, including China, only a few strains have had their full-length genomes sequenced [22].
In this study, the complete nucleotide sequence of a novel GZ2010-L87 strain belonging to GII.4-2009 genocluster was analyzed to establish phylogenetic and evolutionary relationships with other related viral strains circulating worldwide at different time periods.
Materials and methods
Clinical samples
In a NoV surveillance study conducted in patients with sporadic gastroenteritis during winter in South China in 2010, a novel GII.4 variant was identified based on full-length ORF2 and partial RNA-dependent RNA polymerase region sequences. After collection, 10 % (w/v) fecal specimens were homogenized in phosphate-buffered saline (pH 7.3, diethylpyrocarbonate-treated) and centrifuged at 12,000×g for 10 min at 4 °C. Aliquots (1 ml/sample) of the supernatant were kept at −80 °C until analysis.
RNA extraction
Viral RNA was extracted from 140 μl fecal suspensions using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. The RNA pellet was resuspended in 50 μl DNase-free and RNase-free water and was immediately subjected to reverse transcription.
NoV genome amplification
Amplification of the complete NoV strain genome was carried out with the extracted viral RNA as template according to the three-fragment clone protocol [23]. The first two fragments were obtained by one-step reverse transcription PCR (RT-PCR) (TAKARA, Japan), following the manufacturer’s protocol. Briefly, one-step RT-PCR was performed in a 20 μl reaction mixture, containing 0.8 μl enzyme mix, 10 μl 2× RT-PCR buffer, 1.0 μl of 10.0 mmol l−1 primers, 2.0 μl RNA extract, and 5.2 μl RNase-free water. PCR was then performed with the following cycling profile: 50 °C for 30 min, 94 °C for 3 min, followed by 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 3 min, and a final step at 72 °C for 7 min. The third fragment was characterized by 3′ rapid amplification of cDNA ends. Briefly, oligo dT-Adaptor (M13-M4) (TAKARA, Japan) was used as reverse primer for cDNA synthesis with AMV reverse transcriptase (TAKARA, Japan) in a 10 μl reaction mixture, according to the manufacturer’s instructions. The RT reaction mixture was subsequently PCR-amplified with LA Taq (TAKARA, Japan) using M13-M4 and COG2F primers [24] with the following cycling profile: 30 cycles of 30 s at 94 °C, 30 s at 55 °C, and 3 min at 72 °C, followed by a final extension at 72 °C for 7 min.
Cloning and sequencing
The PCR products with appropriate sizes were analyzed by electrophoresis on 1.0 % agarose gel in tris–acetate–EDTA buffer stained with GoldView. The amplified fragments were gel-extracted and cloned into the pMT19T-simple vector (TAKARA, Japan). The recombinant plasmid was then transformed into Escherichia coli DH5α competent cells (TAKARA, Japan). After confirmation by colony PCR, five or more independent clones were randomly picked to be sequenced by primer walking. Sequencing was performed using standard automated methods on an automated sequencer (454GS FLX, Majorbio Co., Ltd., Shanghai, China). The nucleotide sequence of the GZ2010-L87 strain reported in this study was deposited in GenBank under the accession number JX989074.
Comparative analysis
Similarity searches were carried out using the BLAST search (http://www.ncbi.nlm.nih.gov/BLAST/) utility of the National Center for Biotechnology Information (NCBI) database. ORF positions were verified by the ORF finder of NCBI and compared with the reference sequence. The full genome sequence of GZ2010-L87 and the sequences of other similar strains in the same cluster were compared to investigate the differences among the said sequences at the nucleotide and amino acid levels. The mismatched sequences were verified using Bioedit (version 7.1).
Phylogenetic analysis
Multiple alignment was performed using ClustalX for Windows (version 1.83) with the default parameters set for gap opening and gap extension penalties [25]. The phylogenetic relationship of GZ2010-L87 and the representative NoV strains from each genogroup with the entire sequences and subsequences for ORF1, ORF2, and ORF3 were then assessed using the software MEGA (version 5.05) [26]. The reliability of the different phylogenetic groupings was evaluated by bootstrap test (1,000 replications). All nucleotide sequences were genotyped using the NoV automated online genotyping tool (www.rivm.nl/mpf/norovirus/typingtool) [27].
Reference NoV panel
NoV strains with full genomes were used as references for phylogenetic analysis, and their GenBank accession numbers are as follows: AB039774, AB039778, AB039780, AB042808, AB045603, AB083780, AB187514, AF093797, AF097917, AY228235, AY502023, AY741811, AY772730, DQ078814, DQ456824, EF187497, EU424333, FJ692500, GU991354, GU991355, HM748973, JN595867, L07418, M87661, U07611, and X86557. NoV strains with full genomes that were clustered into GII.4-2009 are as follows: GQ845367, GU445325, HM748972, JN400618, JN400619, JN400621–JN400625, JN595867, JQ613570, JQ613571, JQ613573, JX445164–JX445169, JX459901, JX459902, JX459904, JX846928, KC175380, and KC175382–KC175387. NoV strains with full capsid protein VP1 that were clustered into GII.4-2009 are as follows: AB629944, GQ246801, GQ845345, HM191773, HM625866, HM635103, HQ005293, HQ005294, HQ005296–HQ005299, HQ456345, HQ456346, JQ613526–JQ613528, JQ613530–JQ613551, JQ613553–JQ613561, JQ613563–JQ613566, JX459640, JX459642, JX459643, JX459645–JX459647, JX459649–JX459654, JX459656–JX459659, JX644031, JX644032, and JX644034–JX644039.
Results
The complete genome of the GZ2010-L87 strain
The entire nucleotide sequence of GZ2010-L87 (7,559 bp) was submitted to GenBank with accession number JX989074. BLAST results using the GZ2010-L87 nucleotide sequence as query showed that the top three sequences with highest similarity were New Orleans1805/2009/USA, CGMH23/2010/TW, and AlbertaEI045/2010/CA (query coverage = 99 % and maximum identity = 99 %). Three ORFs were included spanning nucleotides 5–5104, 5085–6707, and 6707–7513, respectively (Fig. 1). ORF1 and ORF2 had an overlap of more than 20 nucleotides (nt 5,085–5,104), whereas ORF2 and ORF3 had a single nucleotide overlap (A at position 6707).

Composition of norovirus GZ2010-L87 strain genome. Nucleotides 5–5104, 5085–6707, and 6707–7513 encode ORFs 1, 2, and 3, respectively. ORFs 1 and 2 share a 20-nt overlap, and ORFs 2 and 3 share a 1-nt overlap
Phylogenetic analysis of the GZ2010-L87 strain
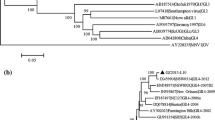
To better understand the genetic relationships between GZ2010-L87 and other completely sequenced NoVs, phylogenetic analysis based on the complete genome with representative strains was performed. Pairwise distances of GZ2010-L87 with other reference strains were between 1.7 % (with New Orleans strain) and 77.4 % (with MNV1 strain). The GZ2010-L87 strain was classified as GII.4 genotype and was most closely related to the New Orleans strain, forming a new cluster with the aforementioned viral strain at the nucleotide level. Genotyping was also confirmed using the NoV genotyping tool (version 1.0) [27], which revealed similar results. Thus, the novel cluster was named GII.4-2009.
To identify the recombination events of the GZ2010-L87 strain, three phylogenetic analyses were also performed using the sequence information for each viral ORF in the 32 completely sequenced NoVs genomes (Fig. 2). GZ2010-L87 was verified to be non-recombinant, forming a cluster with the representative sequence of New Orleans1805/2009/USA based on full-length genome or single ORF sequence analysis [28].

Phylogenetic analyses based on the full genome sequences and three ORFs of representative strains for each NoV genotype: a full genome nucleotide sequences; b ORF1 nucleotide sequences; c ORF2 nucleotide sequences; and d ORF3 nucleotide sequences. Bootstrap values greater than 70 % are shown on the corresponding branches. The scale bar represents the unit for the expected number of substitutions per site. GZ2010-L87 is highlighted with a black diamond
Comparison of GZ2010-L87 genome with other GII.4-2009 strains
The GII.4-2009 variant found recently as a new NoV group has caused infections in various regions including Australia, USA, Europe, and Asia [29–31]. To determine the sequence similarity of GZ2010-L87 with other completely sequenced NoVs belonging to GII.4-2009, the complete nucleotide and deduced amino acid sequences of the GZ2010-L87 strain were compared with those of 31 other completely sequenced NoV strains available from GenBank. Among these strains, 10 were isolated from the USA, 8 from Australia, 7 from Taiwan, and 6 from Canada between 2008 and 2012.
The final pairwise comparison of GZ2010-L87 with the 31 representative NoV GII.4-2009 strains revealed high sequence similarities at the nucleotide level (97.0–99.1 %). Disagreements among the consensus sequence of 31 representative strains were 56 in ORF1, 16 in ORF2, and 14 in ORF3, respectively. Among the different nucleotide sites, 67 belonged to nonsense mutations. The remaining 19 variations resulted in amino acid sequence changes, 9 of which were in ORF1, 3 were in ORF2 (S174, T317), and 7 were in ORF3. The differences of GZ2010-L87 with other representative strains are shown in detail in Table 1, which revealed the rapid evolution of the novel NoV genocluster, particularly in ORF3.
Sequence analysis of the capsid protein sequences of GII.4-2009 strains
NoVs maintained rapid evolution due to the herd immunity, and the antigenicity and receptor-binding function of the new emerging variant would be changed, which mainly embodied in the variation among the viral capsid protein sequence. So, to further explore the diversity and the key mutations of the capsid protein of GII.4-2009, another 76 GII.4-2009 NoV capsid sequences were added to the 31 former representative strains. Multiple sequence alignment of the 108 amino acid sequences revealed 12 sites with the following variations: P174, V231, P294, R297, N341, D357, D372, E376, T377, S393, P396, and I413. Ten of these variations were located in the P2 subdomain (Fig. 3). Multiple sequence alignment was also performed with the consensus sequence obtained from 108 strains and other GII.4 genotypes. A total of 14 sites were found specific to GII.4-2009, namely, S6, V231, P294, S310, T317, T340, N341, D357, S359, R364, A368, D372, N378, P396, I413, and 460Y (Fig. 4). Meanwhile, 12 sites were also found in the hypervariable P2 subdomain. Among all variations, six sites were found as the differences within the GII.4-2009 genocluster as well as the successive GII.4 variant lineages: 231, 294, 341, 357, 372, 413, and these sites maybe still evolved after the new variant emergence. However, all variations described above did not belonged to receptor-binding site 1 (S343, T344, R345, A346, K348, N373, D374, D391, G442, and Y443) which always kept conserved except A346G occurred after 2002 [32, 33]. But some changes (294, 340, 368, 372, and 413) happened in the predicted epitope A (294, 296, 297, 298, 368, 372), B (340, 376), and E (407, 412, 413), of which 294, 372, and 413 were still evolving within the GII.4-2009 genocluster [17, 34]. These mutations occurred in epitope A and E would effected the NoVs immunogenicity [35, 36]. Besides, for most GII.4-2009 NoVs, some variable sites (297, 376, 393, 395) located in epitopes were still conserved as that of the previously circulating dominant variants, but the mutations also existed in a small number of GII.4-2009 strains.

Variation sites in the capsid protein of GII.4-2009 cluster. The sites in P2 region are highlighted in brown above the amino acids, with the N- and C-terminal flanking regions of heterogeneity noted in green for the S domain and blue for P1

Evolution sites of the capsid protein of GII.4-2009 based on the alignment with other GII.4 strains. GII.4-2009 consensus means the consensus sequence of GII.4-2009, which reflected the majority of the amino acids the 108 GII.4-2009 NoV strains. Yellow, amino acid changes during the time; red, changes specific to GII.4-2009. The sites above the amino acids are colored as in Fig. 3. Strain VA387 is included for comparison, as it is a Grimsby-like virus with a solved crystal structure of the P domain
Discussion
NoVs are regarded as the most important cause of acute gastroenteritis worldwide, including China [37–42]. Human NoVs diverge by 45 % in full-length genomes and by 57 % in the VP1 gene [14]. However, no suitable viral reproduction and in vitro culture system is available for the said group. Sequence analysis became more useful for NoV research, particularly when using complete genomic sequences. However, only a few completed NoV genome sequences from China are available in the GenBank [22, 43], which has hindered further research, such as phylogenetic analysis, recombination identification, and evolution research. In this study, the full genome sequence of the GZ2010-L87, a newly isolated strain detected in Guangzhou, was characterized.
In the phylogenetic analysis based on reference NoV strains of different genotypes, GZ2010-L87 was found to belong to NoV GII.4, which is the dominant genotype worldwide. Globally, different GII.4 variants have become the most predominant strains since the mid-1990s [44–47]. Rapid evolution promotes the predominant genotype, with at least seven subclusters occurring every two or three years [48]. The GZ2010-L87 strain was clustered into a new group, with several other strains previously detected in most continents since 2009 [29–31].
In this study, most of the unique amino acid substitutions in the capsid protein of the new strain were identified to be part of the P2 subdomain, which is a hypervariable region. The evolution of P2 by positive selection in response to herd immunity has already been suggested [12]. A recent study indicated that the GII.4 NoV is continuously evolving through the alteration of the surface-exposed receptor-binding domain of the VP1 protein in response to human immune selection [17, 49]. Understanding the viral and cellular factors that make GII.4 and its variants the most prevalent genotype is thus important. Further studies are needed to elucidate the variations in ORF2, which can help in designing NoV vaccines.
In the recent years, limitations in classifying viruses into genogroups based on partial nucleotide sequences have been a growing concern. Thus, analysis of entire nucleotide sequences in NoVs is necessary to improve the accuracy of phylogenetic classification. To date, most whole-genome NoV analyses have been performed in developed countries, such as the USA, Australia, and Japan [21]. This study confirms the GZ2010-L87 strain as the first NoV GII.4-2009 variant isolated in China. The results of this study can be used for future genetic studies on NoVs in China and further research on NoV diversity.
References
B.A. Lopman, A.J. Hall, A.T. Curns, U.D. Parashar, Clin. Infect. Dis. 52, 466–474 (2011)
B.A. Lopman, M.H. Reacher, I.B. Vipond, D. Hill, C. Perry, T. Halladay, D.W. Brown, W.J. Edmunds, J. Sarangi, Emerg. Infect. Dis. 10, 1827–1834 (2004)
M.M. Patel, M.A. Widdowson, R.I. Glass, K. Akazawa, J. Vinje, U.D. Parashar, Emerg. Infect. Dis. 14, 1224–1231 (2008)
J.P. Harris, W.J. Edmunds, R. Pebody, D.W. Brown, B.A. Lopman, Emerg. Infect. Dis. 14, 1546–1552 (2008)
L. Maunula, C.H. von Bonsdorff, Future Virol. 6, 431–438 (2011)
G.P. Richards, C. McLeod, F.S. Le Guyader, Food Environ. Virol. 2, 183–193 (2010)
P.F.M. Teunis, C.L. Moe, P. Liu, S.E. Miller, L. Lindesmith, R.S. Baric, J. Le Pendu, R.L. Calderon, J. Med. Virol. 80, 1468–1476 (2008)
E. Guevremont, J. Brassard, A. Houde, C. Simard, Y.L. Trottier, J. Virol. Methods 134, 130–135 (2006)
P.C. Carling, L.A. Bruno-Murtha, J.K. Griffiths, Clin. Infect. Dis. 49, 1312–1317 (2009)
R.L. Atmar, M.K. Estes, Gastroenterol. Clin. N. 35, 275–290 (2006)
B.V.V. Prasad, M.E. Hardy, T. Dokland, J. Bella, M.G. Rossmann, M.K. Estes, Science 286, 287–290 (1999)
J.J. Siebenga, H. Vennema, B. Renckens, E. Bruin, B.D. van der Veer, R.J. Siezen, M. Koopmans, J. Virol. 81, 9932–9941 (2007)
A. Bertolotti-Ciarlet, S.E. Crawford, A.M. Hutson, M.K. Estes, J. Virol. 77, 11603–11615 (2003)
D.P. Zheng, T. Ando, R.L. Fankhauser, R.S. Beard, R.I. Glass, S.S. Monroe, Virology 346, 312–323 (2006)
T.N. Hoa Tran, E. Trainor, T. Nakagomi, N.A. Cunliffe, O. Nakagomi, J. Clin. Virol. 56, 185–193 (2013)
D.P. Zheng, M.A. Widdowson, R.I. Glass, J. Vinje, J. Clin. Microbiol. 48, 168–177 (2010)
K. Debbink, L.C. Lindesmith, E.F. Donaldson, R.S. Baric, PLoS Pathog. 8, e1002921 (2012)
L.C. Lindesmith, E.F. Donaldson, A.D. Lobue, J.L. Cannon, D.-P. Zheng, J. Vinje, R.S. Baric, PLoS Med. 5, e31 (2008)
G.C. Lee, G.S. Jung, C.H. Lee, Virus Genes 45, 225–236 (2012)
P. Chhabra, A.M. Walimbe, S.D. Chitambar, Infect Genet. Evol. 10, 1101–1109 (2010)
K. Motomura, M. Yokoyama, H. Ode, H. Nakamura, H. Mori, T. Kanda, T. Oka, K. Katayama, M. Noda, T. Tanaka, N. Takeda, H. Sato, N.S.G. Japan, J. Virol. 84, 8085–8097 (2010)
Q. Shen, W. Zhang, S.X. Yang, Y. Chen, T.L. Shan, L. Cui, X.G. Hua, Mol. Biol. Rep. 39, 1275–1281 (2011)
L. Xue, Q. Wu, X. Kou, J. Zhang, W. Guo, J. Appl. Microbiol. (2013). doi:10.1111/jam.12244
T. Kageyama, S. Kojima, M. Shinohara, K. Uchida, S. Fukushi, F.B. Hoshino, N. Takeda, K. Katayama, J. Clin. Microbiol. 41, 1548–1557 (2003)
J.D. Thompson, T.J. Gibson, F. Plewniak, F. Jeanmougin, D.G. Higgins, Nucleic Acids Res. 25, 4876–4882 (1997)
K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei, S. Kumar, Mol. Biol. Evol. 28, 2731–2739 (2011)
A. Kroneman, H. Vennema, K. Deforche, H. von der Avoort, S. Penaranda, S. Oberste, J. Vinje, M. Koopmans, J. Clin. Virol. 51, 121–125 (2011)
E. Vega, L. Barclay, N. Gregoricus, K. Williams, D. Lee, J. Vinje, Emerg. Infect. Dis. 17, 1389–1395 (2011)
C. Yen, M.E. Wikswo, B. Lopman, J. Vinje, U.D. Parashar, A.J. Hall, Clin. Infect. Dis. 53, 568–571 (2011)
G.E. Greening, J. Hewitt, M. Rivera-Aban, D. Croucher, J. Med. Virol. 84, 1449–1458 (2012)
L. Puustinen, V. Blazevic, M. Salminen, M. Hamalainen, S. Rasanen, T. Vesikari, Scand. J. Infect. Dis. 43, 804–808 (2011)
S. Shanker, J.M. Choi, B. Sankaran, R.L. Atmar, M.K. Estes, B.V.V. Prasad, J. Virol. 85, 8635–8645 (2011)
S. Cao, Z.Y. Lou, M. Tan, Y.T. Chen, Y.J. Liu, Z.S. Zhang, X.J.C. Zhang, X. Jiang, X.M. Li, Z.H. Rao, J. Virol. 81, 5949–5957 (2007)
R.S. Baric, E.F. Donaldson, L.C. Lindesmith, A.D. Lobue, Immunol. Rev. 225, 190–211 (2008)
L.C. Lindesmith, V. Costantini, J. Swanstrom, K. Debbink, E.F. Donaldson, J. Vinjé, R.S. Baric, J. Virol. 87, 2803–2813 (2013)
K. Debbink, E.F. Donaldson, L.C. Lindesmith, R.S. Baric, J. Virol. 86, 1214–1226 (2012)
Z.J. Duan, Y. Jin, W.X. Cheng, X.M. Yang, M. Jin, Q. Zhang, Z.Q. Xu, J.M. Yu, L. Zhu, S.H. Yang, N. Liu, S.X. Cui, Z.Y. Fang, J. Clin. Virol. 44, 238–241 (2009)
H.Y. Li, M. Jin, Q. Zhang, N. Liu, S.X. Cui, Z.Y. Fang, Z.J. Duan, J. Infect. 59, 215–218 (2009)
L. Guo, J.D. Song, X.W. Xu, L.L. Ren, J.G. Li, H.L. Zhou, M. Wang, J.G. Qu, J.W. Wang, T. Hung, J. Clin. Virol. 44, 94–98 (2009)
Q. Shen, W. Zhang, S. Yang, Y. Chen, H. Ning, T. Shan, J. Liu, Z. Yang, L. Cui, J. Zhu, X. Hua, Arch. Virol. 154, 1625–1630 (2009)
Y. Gao, M. Jin, X. Cong, Z.J. Duan, H.Y. Li, X.L. Gun, Y. Zuo, Y.M. Zhang, Y. Zhang, L. Wei, J. Med. Virol. 83, 1078–1085 (2011)
M. Zeng, Z.X. Gong, Y.X. Zhang, Q.R. Zhu, X.H. Wang, Jpn. J. Infect. Dis. 64, 417–422 (2011)
M. Jin, H.P. Xie, Z.J. Duan, N. Liu, Q. Zhang, B.S. Wu, H.Y. Li, W.X. Cheng, S.H. Yang, J.M. Yu, Z.Q. Xu, S.X. Cui, L. Zhu, M. Tan, X. Jiang, Z.Y. Fang, J. Med. Virol. 80, 1997–2004 (2008)
J.S. Noel, R.L. Fankhauser, T. Ando, S.S. Monroe, R.I. Glass, J. Infect. Dis. 179, 1334–1344 (1999)
B. Lopman, H. Vennema, E. Kohli, P. Pothier, A. Sanchez, A. Negredo, J. Buesa, E. Schreier, M. Reacher, D. Brown, J. Gray, M. Iturriza, C. Gallimore, B. Bottiger, K.O. Hedlund, M. Torven, C.H. von Bonsdorff, L. Maunula, M. Poljsak-Prijatelj, J. Zimsek, G. Reuter, G. Szucs, B. Melegh, L. Svennson, Y. van Duijnhoven, M. Koopmans, Lancet 363, 682–688 (2004)
M.A. Widdowson, E.H. Cramer, L. Hadley, J.S. Bresee, R.S. Beard, S.N. Bulens, M. Charles, W. Chege, E. Isakbaeva, J.G. Wright, E. Mintz, D. Forney, J. Massey, R.I. Glass, S.S. Monroe, J. Infect. Dis. 190, 27–36 (2004)
R.A. Bull, E.T.V. Tu, C.J. McIver, W.D. Rawlinson, P.A. White, J. Clin. Microbiol. 44, 327–333 (2006)
X.L. Pang, J.K. Preiksaitis, S. Wong, V. Li, B.E. Lee, PLoS ONE 5, e11599 (2010)
R.A. Bull, P.A. White, Trends Microbiol. 19, 233–240 (2011)
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31271878) and the National Key Technology Research and Development Program (2012BAK08B07).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xue, L., Wu, Q., Kou, X. et al. Complete genome analysis of a novel norovirus GII.4 variant identified in China. Virus Genes 47, 228–234 (2013). https://doi.org/10.1007/s11262-013-0945-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-013-0945-8