
Abstract
Apple mosaic virus (ApMV) is a widespread ssRNA virus which infects diverse species of Rosales. The phylogenetic analysis of complete capsid protein gene of the largest set of ApMV isolates discriminated two main clusters of isolates: one cluster correlates with Maloideae hosts and Trebouxia lichen algae hosts; a second with hop, Prunus, and other woody tree hosts. No correlation was found between clusters and geographic origin of virus isolates, and positive selection hypothesis in distinct hosts was not confirmed: in all virus populations, purifying selection had occurred. GGT→AAT substitution resulted in Gly→Asn change inside the zinc-finger motif in the capsid protein was revealed specific for discrimination of the clusters and we hypothesise that could influence the host preference.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apple mosaic virus (ApMV), first reported in Rosa spp. and Malus domestica in USA [1], is a species in the Ilarvirus genus subgroup III (Bromoviridae family) [2]. In addition to the roses (Rosa sp.), ApMV infects a number of woody plants including blackberry (Rubus), raspberry (R. idaeus), black raspberry (R. occidentalis), red current (Ribes rubrum), apple and crab apple trees (Malus pumila, M. silvestris), apricot, cherry, almond, plum, and peach (Prunus sp.), and mountain ash (Sorbus aucuparia) (all from the family Rosaceae); hazelnut (Corylus avellana), silver birch (Betula pendula), and chestnuts (Aesculus hippocastanum, A. × carnea, A. flava, A. parviflora) (all belonging to the order Fagales); and hop (Humulus lupulus, family Cannabaceae), [3]. One isolate is known from strawberry (Fragaria sp.) [4], and six isolates were found in lichen symbiotic Trebouxia algae [5]. The leaves of infected trees show typical chrome-yellow line pattern, bright yellow blotches and vein clearing, rings, and oak-leaf pattern, which appear at the beginning of summer [6]. The symptomatic leaves often drop prematurely. The symptomatology is generally not of diagnostic significance, because similar symptoms may be produced also by other Ilarviruses [6]. In hop, leaves of sensitive cultivars show a light green or yellow pattern which could become necrotic, although, many insensitive cultivars show no symptoms of virus infection [3, 7]. ApMV is a mechanically and graft-transmissible pathogen that is present worldwide and persists in propagative material, which is probably the main source of infection by the virus. It is not pollen-borne, nor does it occur in seedling rootstocks. It has not been identified in naturally infected weeds, and the source of the virus remains unknown [8]. In hops, ApMV infection decreases bitter alpha acid yield by 5–34 % [7]. In pome fruits, the infection cause the retardation of growth and premature drop of leaves, in stone fruits, it may cause growth reduction and yield losses, in some sensitive almond cultivars the virus induce failure of blossom and leaf failure symptom [9].
Studies on potyviruses have shown that the extant populations of different species are only decades to centuries old, reflecting an evolutionary burst related to the intensification of agricultural practices, colonization of new areas by Europeans, and post-Columbian world trade over the last 500 years [10]. Previous sequence analyses of ApMV performed on a limited number of strains revealed significant similarity among the isolates from distinct hosts like hop, apples, and stone-fruit plants [7, 11–13]. An unusually high-sequence identity of isolates from geographically distant localities points to a hypothesis of host-conditioned modification and a fixation of nucleotide substitutions in fruit hosts from the subfamily Maloideae in contrast to hop, Prunus, and other host species of ApMV. In addition to the effects of the host, an accidental transfer of the virus from nonrelated hosts within a locality could also contribute [11]. The source of virus inside lichen algae is completely unknown, since four were from lichens growing on tree bark and two from lichens growing on ground [5]. Adaptation to the host species is of fundamental importance, thereby, elevating the reproductive rate of the virus above the critical value needed for sustained transmission [14].
Molecular evolution of viruses is driven by both selection and recombination. The fixation rate of positive ssRNA viruses is in the range of 10−2–10−4 mutations/site/year [15]. However, strong bottlenecks existing for viruses during migration between different hosts or between plants of a given host via a vector [14] is accomplished in Ilarviruses due to the fact that no insect vector is known to transmit them. Therefore, another genetic bottleneck like selection during virus movement in plants can be more important here [16]. In Ilarviruses, the coat protein (CP) is generally required for systemic movement and may be required for cell-to-cell spread [17]. Crucial for the proper CP functions is its RNA-binding and protein-binding activity. Highly conserved arginine in Q/K/R·P/N·T·X·R·S·R/Q·Q/N/S·W/F/Y·A binding consensus sequence and a zinc-finger motif (C·x2·C·x·h·x·H·x3·c·x2·C·x2·C·H/C) have been recognized in N-terminal part of CP of several Ilarviruses [18–20].
In ApMV, the consensus CP sequence has been established as having 654 nt, but isolates with insertions 6–15 nt after nt position 141 have been described [12]. Also, isolates from apple, pear, and cherry trees are known with CP genes encoding proteins 221, 222, and 223 aa long respectively. Another length variant is known from almond, with a CP 220 aa long as well as variants from hop, prune, and mahaleb which are 218 aa long [11–13, 21–25]. A stringent and robust criterion for detecting adaptive evolution in a protein-coding gene is an accelerated nonsynonymous (Pi(a), amino acid replacing) rate relative to the synonymous (Pi(s), silent) rate of substitutions, with the rate ratio Pi(a)/Pi(s) > 1. As silent mutations do not change the amino acid whereas replacement mutations do, the differences in their fixation rates provides a measure of selective pressure on the protein. In this study, we analyzed CP sequences of 65 ApMV isolates, including 23 new sequences from hop and new sequences from mountain ash, hazelnut, peach, and apricot, to determine the molecular variability of ApMV and examine the correlations between amino acid substitutions and host species or geographic origin.
Materials and methods
Virus isolates
Twenty-three ApMV isolates from asymptomatic hop plants originating from different locations were obtained from the collection kept at the Hop Research Institute Co., Ltd. (Žatec, Czech Republic). Isolates from mountain ash (Sambucus nigra), hazelnut (Corylus avellana), peach (Prunus persica), and apricot (Prunus armeniaca) originated from the Czech Republic and Italy (Table 1).
Nucleic acid isolation and reverse transcription
Infected leaves from plants growing either in a screen house or in the wild were used in this study. The nucleic acid was isolated from 100 mg of plant material using TriPure Isolation Reagent (Roche), then eluted to obtain 80 μl of analyte. Reverse transcription was performed from 750 ng of total RNA in a 50 μl mixture containing 1× First-Strand Buffer (Invitrogen), 0.5 μg random hexamers (Roche Diagnostics), 0.5 mM dNTP, 40 U RiboLock™ RNase Inhibitor (Fermentas), 4 mM DTT (Invitrogen), and 40 U MuMLV Reverse Transcriptase (Invitrogen) for 55 min at 42 °C.
Amplification and sequencing
Amplification was performed with TaKaRa Ex Taq™ polymerase (Takara) using primers 5′-GGCCATTAGCGACGATTAGTC-3′ and 5′-ATGCTTTAGTTTCCTCTCGG-3′ amplifying the complete CP gene localized within the positions 1126–1794 nt [9]. Cycling parameters were as follows: denaturation at 94 °C for 30 s, annealing at 57 °C for 30 s, and extension at 72 °C for 45 s, in 35 cycles, with a final extension at 72 °C for 7 min. PCR products were purified using MiniElute Gel Extraction Kit (Qiagen), ligated into the pSC-A vector, and transformed to competent cells. Recombinant plasmids harboring a cDNA insert were sequenced in both orientations using M13 primers (Macrogen, the Netherlands).
Data analysis
The resulting sequences were analyzed using the BioEdit 7.0.9 program (Ibis Biosciences, USA) and deposited in GenBank under accession numbers HE866936 to HE866962. An additional complete CP sequences published previously were used for the analyses (Table 1). The CP sequence of PNRSV (NC_004364) was used as an outgroup.
Multiple alignments
Multiple alignments were carried out using the Clustal X, version 2.0 [26] with gap opening penalty set to 15 and gap extension penalty set to 6.66.
Recombination
The possible occurrence of recombination was tested using the default conditions of the suite of programs included in RDP3 Beta41 [27].
Phylogenetic analysis
MEGA 5 was used to find the best DNA model for our data [28]. The optimal substitution pattern was determined according to the program’s Bayesian information criterion, which indicated as best the Kimura 2-parameter model with discrete gamma distribution (+G), and five rate categories or the Kimura 2-parameter model as just stated but assuming that a certain fraction of sites are evolutionarily invariable (+I).
Substitution rates and evolution
Synonymous and nonsynonymous substitution analyses were also conducted in DnaSP v5 [29] according to the Nei–Gojobori model and using the Jukes–Cantor correction as well as for carrying out the neutrality test.
Results and discussion
Sequence analysis of CP gene of ApMV isolates
The CPs of all the analyzed ApMV isolates from hop cultivars as well as from non-hop hosts P. armeniaca, P. persica, S. nigra, and C. avellana consisted of 654 nt (218 aa) residues. No heterogeneity in the gene size was noticed.
No recombination event was detected among the 65 sequences by more than two methods carried out in RDP3 beta 41 and with window size set at 25 nt.
Neighbor-joining (NJ) and maximum likelihood (ML) phylogenetic analyses of this largest set of sequences to date were compared to test the occurrence of significant differences, but the tree topologies were highly similar. Therefore, only those results obtained with the ML method are shown here.
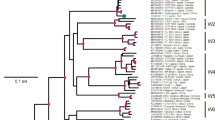
The ApMV capsid protein gene is monophyletic for all the isolates known today. Phylogenetic analyses of the nucleotide sequences indicated two main clusters of isolates and two single-standing isolates from almond (PAM-ITA) and from unknown Chinese host (XX1-CHN) supported by bootstrap values above 50. Cluster I is more homogeneous than cluster II. It contained all hop isolates irrespective of geographic origin, isolates from various Prunus hosts, an isolate from hazelnut (CA-CZE), and one isolate from pear (PC1-CZE). Cluster II contained all the remaining isolates from apple and pears, one isolate from Prunus avium (PAV-IND), and isolates from lichen Trebouxia algae symbionts. We can speculate that these extra-host standing PC1-CZE and PAV-IND isolates are accidental infection of pear and Prunus avium, respectively, that originated from another primary host growing in their vicinities (blackthorn in case of PC1-CZE and apple in case of PAV-IND) (Petrzik unpublished; [13]). Subclusters Ia, Ib, Ic, IIa, and IIb supported by high-bootstrap values are recognizable on the tree (Fig. 1). The nature of subclusters Ib and Ic, which contained two isolates each, is supported by clustering with another 8 and 9 hop isolates, respectively, when 399 nt long central part CP of larger set of isolates was used for phylogenetic analysis (Supplementary Table 1). In this analysis, new cluster III contained isolate from strawberry and two Chinese isolates from unknown hosts occurred (Supplementary Figure 1).

Clusters of ApMV isolates based on the maximum likelihood phylogenetic tree. Bootstrap values was applied using 1000 replicates
Mean codon-based evolutionary diversity characteristics (Pi(S), Pi(a)) were computed for complete CP gene in 65 sequences; clusters I, and II; and also for subclusters Ia, IIa, and IIb (Table 2). The mean Pi(a)/Pi(s) ratio was 0.178 and is close to 0.187 value computed for smaller amount of isolates previously [30]. Substitution rates, smaller than 1, were found in all clusters and subclusters here. This suggests that, in all virus populations, purifying selection occurred due to natural selection, which conserved the protein sequences despite the occurrence of nucleotide polymorphisms. The Pi(a)/Pi(s) ratio of isolates from subcluster IIa only is significantly higher than that of the other subclusters, thus, indicating that those isolates are under tighter functional constraints. In subcluster IIa, there are isolates from apple hosts from different geographic origin, but where shift mutations in the CP gene occur [12, 21]. Similar difference between groups of isolates had been observed in comparison of nucleotide diversity for CP gene of almond and “other hosts” isolates of the related Prune dwarf virus (Ilarvirus): a significantly higher proportion of nonsynonymous substitutions had been found for the cluster of almond variants, than for the “other hosts” isolates. The differences in selection pressure had been explained by the agricultural practice here [31].
Detailed aa sequence comparison revealed unique motif characteristic for the clusters (Supplementary Figure 2): The position Gly7 in the context MVCKYCGHT is specific for all the sequences from cluster I (and for PAM-ITA and XX1-CHN isolates); Asn7 is specific for sequences from cluster II in the same position (supposing presence of corresponding motives in all Australian, Indian, and Latvian isolates, also, where the 5′-end of CP was not sequenced). The Gly7/Asn7 position is in immediate proximity with the zinc-finger motif and could influence its nucleic acid- and/or protein-binding activity [32] and indirectly the host preference. This change is also of some value for discrimination, as it is in a putative antigenic site on the CP [12].
The positive selection hypothesis distinctly for the apple mosaic virus CP gene has not been confirmed. Significant differences in substitution rates in isolates from distinct host-related clades were not observed, however, some groups of isolates (especially that with frame-shift mutations from subcluster IIa) showed much higher values of mean diversity suggesting different functional constraints comparing the rest of isolates (results not shown). It is highly probable that finer classification of cluster II will be performed in future with the growing number of sequences from new hosts.
References
R.P. White, Plant Dis. Rep. 12, 33–34 (1928)
J. Bujarski, M. Figlerowicz, D. Gallitelli, M.J. Roossinck, S.W. Scott, Genus Ilarvirus. in Virus taxonomy, Classification and nomenclature of viruses, eds by A.M.Q. King, M.J. Adams, E.B. Carstens, E.J. Lefkowitz. Ninth Report of the International Committee on Taxonomy of Viruses (Elsevier Academic Press, Amsterdam, 2012) pp. 972–975
CAB International, Crop Protection Compendium (CAB International, Wallingford, 2003)
I.E. Tzanetakis, R. Martin, Plant Dis. 89, 431 (2005)
K. Petrzik, J. Vondrák, M. Barták, O. Peksa, O. Kubešová, Eur. J. Plant Pathol. (2013, in press)
S. Paunovic, G. Pasquini, M. Barba, Apple mosaic virus in stone fruits, in Virus and virus-like diseases of pome and stone fruits, ed. by A. Hadidi, M. Barba, T. Candresse, W. Jelkmann (APS Press, St. Paul, 2011), pp. 91–95
D.R. Crowle, S.J. Pethybridge, G.W. Leggett, L.J. Sherriff, C.R. Wilson, Plant Pathol. 52, 655–662 (2003)
R.W. Fulton, Apple mosaic virus. CMI/AAB Descriptions of plant viruses, 83 (Association of Applied Biologists, Wellesbourne, 1972)
A. Hadidi, M. Barba, Economic impact of pome and stone fruit viruses and viroids, in Virus and virus-like diseases of pome and stone fruits, ed. by A. Hadidi, M. Barba, T. Candresse, W. Jelkmann (APS Press, St. Paul, 2011), pp. 1–7
A.J. Gibbs, D. Fargette, F. García-Arenal, M.J. Gibbs, J. Gen. Virol. 91, 13–22 (2010)
K. Petrzik, Eur. J. Plant Pathol. 111, 355–360 (2005)
K. Petrzik, O. Lenz, Arch. Virol. 147, 1275–1285 (2002)
V. Lakshmi, V. Hallan, R. Ram, N. Ahmed, A.A. Zaidi, A. Varma, Indian J. Virol. 22, 44–49 (2011)
C. Desbiez, B. Moury, H. Lecoq, Infect. Genet. Evol. 11, 812–824 (2011)
R. Sanjuán, PLoS Pathog. 8(5), e1002685 (2012)
S.F. Elena, P. Agudelo-Romero, P. Carrasco, F.N. Codoner, S. Martín, C. Torres-Barceló, R. Sanjuán, Heredity 100, 478–483 (2008)
E.A.G. van der Vossen, L. Neeleman, J.F. Bol, Virology 202, 891–903 (1994)
P. Ansel-McKinney, S.W. Scott, M. Swanson, X. Ge, L. Gehrke, EMBO J. 15, 5077–5084 (1996)
P.C. Sehnke, A.M. Mason, S.J. Hood, R.M. Lister, J.E. Johnson, Virology 168, 48–56 (1989)
J.A. Sánchez-Navarro, V. Pallás, J. Gen. Virol. 75, 1441–1445 (1994)
R.H. Alrefai, P.J. Shiel, L.L. Domier, C.J. D’Arcy, P.H. Berger, S.S. Korban, J. Gen. Virol. 75, 2847–2850 (1994)
D. Guo, E. Maiss, G. Adam, R. Casper, J. Gen. Virol. 76, 1073–1079 (1995)
P.J. Shiel, R.H. Alrefai, L.L. Domier, S.S. Korban, P.H. Berger, Arch. Virol. 140, 1247–1256 (1995)
G.P. Lee, K.H. Ryu, H.R. Kim, C.S. Kim, D.W. Lee, J.S. Kim, Plant Pathol. J. 18, 259–265 (2002)
T. Thokchom, T. Rana, V. Hallan, R. Ram, A.A. Zaidi, Phytoparasitica 37, 375–379 (2009)
M.A. Larkin, G. Blackshields, N.P. Brown, R. Chenna, P.A. McGettigan, H. McWilliam, F. Valentin, I.M. Wallace, A. Wilm, R. Lopez, J.D. Thompson, T.J. Gibson, D.G. Higgins, Bioinformatics 23, 2947–2948 (2007)
D. Martin, P. Lemey, M. Lott, V. Moulton, D. Posada, P. Lefeuvre, Bioinformatics 26, 2462–2463 (2010)
K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei, S. Kumar, Mol. Biol. Evol. 28, 2731–2739 (2011)
P. Librado, J. Rozas, Bioinformatics 25, 1451–1452 (2009)
E.R. Chare, E.S. Holmes, J. Gen. Virol. 85, 3149–3157 (2004)
F. Fonseca, J.D. Neto, V. Martins, G. Nolasco, Arch. Virol. 150, 1607–1619 (2005)
S.S. Krishna, I. Majumdar, N.V. Grishin, Nucleic Acids Res. 31, 532–550 (2003)
S.W. Scott, M.T. Zimmermann, X. Ge, D.J. MacKenzie, Eur. J. Plant Pathol. 104, 155-161 (1998)
Acknowledgments
The authors thank Dr. Donato Boscia from Istituto di Virologia Vegetale CNR, UOS di Bari, Italy, for providing the infected material. This research was supported by S Grant from the Ministry of Education, Youth and Sports of the Czech Republic and Project No. MSM6046070901.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Apple mosaic virus isolates with partial CP sequences. (DOC 64 kb)
Fig. S1
Clusters of 91 ApMV isolates based on the maximum likelihoodphylogenetic tree of 399 nt long central part of CP. Bootstrap values was applied using 1000 replicates. Asterisk (*) marks partial CP sequences. (DOC 151 kb)
Fig. S1
Amino acid alignment of selected isolates. The position of distinctive Gly7 in the zinc-finger motif is highlighted. (PDF 219 kb)
Rights and permissions
About this article
Cite this article
Grimová, L., Winkowska, L., Ryšánek, P. et al. Reflects the coat protein variability of apple mosaic virus host preference?. Virus Genes 47, 119–125 (2013). https://doi.org/10.1007/s11262-013-0925-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-013-0925-z