
Abstract
Norovirus (NoV) is the major etiological agent causing foodborne and waterborne outbreaks worldwide. We developed a novel duplex real-time quantitative RT-PCR assay designed for the simultaneous detection of and discrimination between NoV genogroups GI and GII, by targeting the short junction region between ORF1 and ORF2, with sensitivity and efficiency comparable to those of each simplex RT-PCR assay. This new duplex assay was evaluated against clinical stool (n = 82) and environmental (groundwater or surface water, n = 60) specimens from South Korea, and the results were compared with those of conventional RT-PCR (cRT-PCR) assays. The duplex assay detected more positive samples than did the cRT-PCR for both clinical (74 vs. 71) and, more strikingly, environmental (24 vs. 10) specimens. No cross-reactivity against specimens containing other enteric viruses such as rotavirus, adenovirus, and poliovirus were observed. These results suggest that this newly developed duplex real-time RT-PCR assay can be used for the sensitive and simultaneous genogroup-specific detection of NoV in both clinical and environmental specimens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Norovirus (NoV), which was previously known as small round structured virus or Norwalk-like virus, is the leading etiological agent of acute nonbacterial gastroenteritis in both developing and developed countries worldwide [1]. Its virion is a 30 to 40-nm non-enveloped icosahedral particle that contains a positive-sense, single-stranded RNA genome of approximately 7.5–7.7 kb. The NoV genome consists of three open reading frames: ORF1 encodes several non-structural proteins, RNA-dependent RNA polymerase (RdRp), and ORF2 and ORF3 encode the major and the minor capsid proteins, respectively. Based on the nucleic acid sequences of RdRp and the capsid genes, NoVs are currently classified into five different genogroups (GI through GV), with GI and GII found predominantly in humans [2, 3].
NoV is an important foodborne and waterborne pathogen, and it causes acute gastrointestinal illness. About 10–100 particles of this virus can induce diarrhea, abdominal pain, vomiting, and even death [4, 5]. Such symptoms lead to significant viral amplification of about 100 million particles per 1 ml of feces or vomit [6]. Therefore, much attention has to be paid to this virus due to the large outbreaks of the disease when water or food is contaminated with very small amount of NoV. As a result, NoV was listed on the Contaminant Candidate List 3 (CCL3) announced in 2009 by the United States Environmental Protection Agency (US EPA). In South Korea, the largest foodborne outbreak was caused by NoV in metropolitan Seoul and Gyeonggi area in 2006 by the Korea Food and Drug Administration (KFDA).
NoV is not cultivable in routine cell cultures, hence, molecular methods such as conventional reverse transcriptase-PCR (cRT-PCR) and real-time quantitative RT-PCR (qRT-PCR) are the most sensitive and accurate, and hence, the most widely used ones for NoV detection [7–11]. Methods like EM or enzyme immunoassays were used at one time, but they are not comparable to the RT-PCR-based methods in terms of accuracy and sensitivity, which is perhaps attributable to the very small size and high antigenic variability of NoV [12–14]. Temporal variation of the sequence of RNA viruses has also been reported [15, 16], and NoV isolates display sequence variation, even in the same geographical region of a NoV outbreak [17]. Therefore, RT-PCR-based methods can be more effective, as long as the target sequences are designed specifically at the non-coding junction region between ORF1 and ORF2, which is known to be one of the most strongly conserved regions of NoV [18].
This study describes the development and optimization of a duplex qRT-PCR assay for broadly reactive genogroup-specific and sensitive detection of NoV. This assay increased the efficiency toward GI by introducing a minor groove binder (MGB) to the NoV GI type probe [19, 20]. None of the published NoV assays has been optimized for duplex qRT-PCR assay without loss of the sensitivity and efficiency of each simplex assay. Furthermore, a direct comparison with the two types of cRT-PCR assays was performed, using 142 fecal and environmental samples to validate the efficiency and sensitivity of this duplex qRT-PCR assay.
Materials and methods
Preparation of NoV RNA samples
NoV isolates were obtained from fecal and environmental samples collected in 2008 and were subsequently genotyped by PCR amplification and nucleic acid sequencing. The two types of cRT-PCR sets (GSK and SR sets) were exploited for the detection of NoV GI and GII groups. Genotyping was performed by BLAST and phylogenetic analysis after nucleotide sequencing of the PCR amplicons as described below.
Eighty-two human acute gastroenteritis stool samples were obtained from the Seoul Institute of Public Health and Environment (http://sihe.seoul.go.kr), Gwang-ju Health and Environment Research Institute (http://hevi.gjcity.net), Catholic Medical Center (http://www.cmc.or.kr), and Seoul National University Hospital (http://www.snuh.org) in 2008. Sixty environmental samples from underground and river water sources in the 2008 winter season were concentrated by NanoCeram cartridge filter (5 inches, Argonide, USA) as described in a previous study [21]. Stool samples were prepared as 10% suspensions in phosphate-buffered saline (pH 7.4) and centrifuged at 4°C for 10 min at 1,000×g, and then the supernatant was resuspended in 10% chloroform and re-centrifuged. These supernatants and the concentrated environmental samples were subjected to total RNA extraction using the QIAamp viral RNA mini kit (Qiagen, Germany) according to the manufacturer’s instructions. The 142 eluted RNA samples were aliquoted and used for NoV detection assays. A negative control (elution buffer) was included for each experiment.
cRT-PCR assays
RNA samples (2.5 μl) were subjected to cRT-PCR and semi-nested cRT-PCR using GSK and SR primer sets specific for GI [G1SKF and G1SKR {amplification product size (aps): 329 base pair (bp)}; SRI-1, SRI-2, and SRI-3 (aps: 241 bp)] and GII [G2SKF and G2SKR (aps: 343 bp); SRII-1, SRII-2, and SRII-3 (aps: 203 bp)], as described elsewhere [22, 23]. cRT-PCR (hereafter, Kojima assay) and nested cRT-PCR (hereafter, Boxman assay) were carried out using the OneStep RT-PCR kit (Qiagen) and AccuPower HotStart PCR PreMix kit (Bioneer, Korea), respectively, according to the manufacturer’s instructions. The products were analyzed on a 1% agarose gel.
Nucleic acid sequencing and phylogenetic analysis
The products from the cRT-PCR assays were extracted from the agarose gel and purified using the QIAquick gel extraction kit (Qiagen), and the nucleotide sequences were determined using a 3730xl DNA analyzer (Applied Biosystems) by Cosmogene-tech (Korea). The nucleic acid sequences from this study were submitted to GenBank (accession numbers JF896115–JF896196). Genotyping was based on nucleotide sequence comparisons and multiple sequence alignments that were performed using the BLAST program (NCBI), ClustalW tool in the MegAlign program (Lasergene 6, USA), and then neighbor-joining method provided in the Molecular Evolutionary Genetic Analysis software package (MEGA version 5). A total of 82 NoV isolates were genotyped in this study (Table 1; Fig. 1).

Phylogenetic tree of NoV sequences examined and generated in this study. Phylogenetic tree based on nucleotide sequences of 11 GI NoV samples with 6 GI reference strains, and 71 GII NoV samples with 13 GII reference strains [37]. The strains names are marked in abbreviation letters; the front letter (S stool, E environment), the following letter (S Seoul Institute of Public Health and Environment, G Gwang-ju Health and Environment Research Institute, K Catholic Medical Center, D and O Seoul National University Hospital, R river water, U underground water). NoV genogroups and genotypes identified in this study are indicated in parenthesis. The scale bar represents 0.05 substitutions per nucleotide position. Bootstrap values of 1,000 repeated analyses are indicated for the major nodes
Preparation of RNA transcript standards
In vitro-transcribed artificial NoV RNA templates were used as NoV GI and GII qRT-PCR standards, as described previously [24]. The pGEM-T easy-derived constructs containing the cRT-PCR product of either NoV GI-1 (Genbank accession number: FJ384783) or NoV GII-4 (Genbank accession number: AB541262) were linearized and then subjected to in vitro transcription using the MAXIscript SP6/T7 kit (Ambion, USA) to generate artificial NoV GI and GII RNA transcripts. Tenfold serial dilutions were made for each transcript, and these were subjected to qRT-PCR in both simplex and duplex formats to determine the efficiency and theoretical sensitivity based on the standard curves.
Design of probe and primer sets for duplex qRT-PCR
Thirty-five NoV sequences from GenBank for GI (Genbank accession number: M87661, L07418, U04469, AY502016, AY502007, AY502008, AB187514, AB042808, AF093797, NC001959, AB039774, AB081723, AB187514, L23828, AF414403, AF414405, AY038598, AJ313030, AF414402, AF414404, AF414406, AF093797) and GII (X86557, X81879, U07611, U22498, X76716, AF145896, AF190817, AB039775, AB039776, AB039778, AB039780, AB039781, AB039782) were aligned using the ClustalW tool in the MegAlign program. The probe and primer sequences were designed based on the sequence alignment results, and were tested for their theoretical properties including melting temperatures, hairpin/dimer formation, self-annealing sites, and probe–probe/primer–primer/primer–probe interactions using the PrimerSelect program (Lasergene 6) and Primer Express 3.0 software (Applied Biosystems). The probe and primer sets used are listed in Table 2.
Duplex and simplex qRT-PCR assays
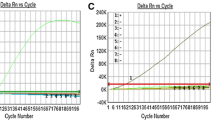
Duplex and simplex qRT-PCR assays were carried out in a 25-μl reaction mixture containing 2.5 μl of extracted RNA, 200 nM of each primer (NKP1F and NKP1R for GI and/or NKP2F and NKP2R for GII), 100 nM of each probe (NKP1P for GI and/or RING2-TP for GII), 12.5 μl of 2× RT-PCR buffer, and 1 μl of 25× RT-PCR enzyme mixture using a AgPath-ID One-Step RT-PCR kit (Ambion) according to the manufacturer’s recommendations. PCR amplification was performed with a 7300 Real-Time PCR System (Applied Biosystems) under the following conditions: RT for 10 min at 42°C, initial denaturation at 95°C for 10 min, followed by 45 cycles of amplification with denaturation at 95°C for 15 s and annealing–extension at 60°C for 60 s. Fluorescence data, in the VIC channel for GI and in the FAM channel for GII, were collected during each annealing–extension step in one tube and analyzed with 7000 SDS 1.1 RQ Software (Applied Biosystems). Negative controls were included in each analytical procedure. The cycle threshold (CT) was plotted against log (RNA amount per reaction) to determine the slope of each curve by linear regression analysis. The positivity of detection was determined with cutoff CT values of 43 and 42 for GI and GII, respectively, based on the standard curves. The reaction efficiency (E) under our experimental conditions was calculated with the formula E = 10−1/slope, the theoretical limit of which is 2 [25]. For the samples with high CT values, the experiment was performed in duplicate. All of these experiments were consistent with each other.
Statistical analysis
Pearson’s χ2 test of independence based on 2 × 2 contingency tables was used to test for the statistical significance of the difference between the GI and GII distribution in clinical and environmental samples.
Results
Conventional RT-PCR and genotyping of NoV isolates
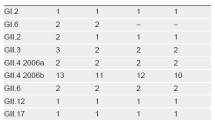
NoV isolates were obtained from fecal and environmental samples collected in 2008 and were subsequently genotyped by PCR amplification and nucleic acid sequencing. The two types of cRT-PCR sets (GSK and SR sets) were exploited for the detection of NoV GI and GII groups, based on that cRT-PCR assay using GSK primers is the standardized NoV detection protocol recommended by the Korean Center for Disease Control [26]. We genotyped 82 NoV isolates from a total of 142 samples (82 clinical and 60 environmental, Table 1; Fig. 1). Interestingly, the G1SK and G2SK primer sets designed by Kojima et al. [22] were more effective than the SRI and SRII primer sets by Boxman et al. [23] for NoV detection from the samples tested (data not shown). Based on the multiple alignment of the nucleotide sequences from these isolates with a total of 35 NoV sequences covering each genotype, there were 6 GI genotypes (GI-1, GI-5, GI-7, GI-8, GI-9, and GI-14) and 12 GII genotypes (GII-1 to -8, GII-11, GII-13, GII-14, and GII-16). The most frequent genotype in Korea was GII-4 (n = 32), followed by GII-3 (n = 14), which is in accordance with the prevalence of NoV genotypes worldwide [27]. GI isolates were detected less frequently (n = 11), with GI-1 being the most prevalent (n = 5) and detected only in the environmental samples (Table 1). These findings suggest that the Kojima assay may be more appropriate than the Boxman assay for NoV detection, at least in our samples, where the genotype prevalence was similar to that reported previously worldwide, so we could use the collected samples to evaluate our new method, as described below.
Design of primer and probe sets for a duplex qRT-PCR assay
We retrieved 35 representative NoV sequences (22 GI and 13 GII) from GenBank and constructed a multiple alignment using the ClustalW program, revealing a highly conserved region at the ORF1–ORF2 junction for both GI and GII genogroups (data not shown), as described previously [18]. The primer and probe sets were basically adopted from the previous reports [7, 28], with slight modifications based on the higher sequence variation in GI, compared to GII. For GI, we introduced a degenerate nucleotide (Y) in the forward primer, and subsequently, two or three additional nucleotides were inserted at the 5′-end of the reverse primer for appropriate annealing (Table 2). For the detection probe, the MGB quencher, which is known to enhance the thermal stability of the probe binding to the template [29], was exploited due to the fewer successive identical residues in the amplicons in all GI subtypes [7]. For GII, we newly designed the forward primer at 5,012–5,034, which was optimized for the duplex assay for GI and GII considering amplicon sizes, melting temperatures, hairpin/dimer formation, and probe–probe, primer–primer, and primer–probe interactions (data not shown).
Development and optimization of a duplex qRT-PCR assay
Based on the designed primer and probe sets, we then optimized the duplex qRT-PCR assay by first using NoV RNA standards for both GI and GII. The efficiency and sensitivity of the assay were determined using tenfold serial dilutions of in vitro-transcribed RNA templates (i.e., about 1–1010 RNA copies per reaction). A linear regression analysis was performed for each genogroup in duplex and simplex formats. As shown in Table 3, the reaction efficiencies calculated using the slope of each equation for each target in the simplex assays for GI and GII were 1.955 and 1.994, respectively, whereas those values in the duplex assays for GI and GII were 1.972 and 1.993, respectively, indicating the reliable amplification in our experimental setting of both GI and GII in the duplex assay as well. Furthermore, from the y-intercept of the standard curves, the theoretical lower detection limits for GI and GII did not differ significantly between the duplex and simplex assays, indicating no cross-reactivity between GI and GII and no interference in the detection capabilities for GI and GII in the duplex assay, compared to the simplex assays. Overall, the duplex assay displayed better efficiency with comparable sensitivity compared with the previous one developed by Jothikumar et al. [28], at least against the RNA standards under our experimental conditions. Based on our experimental data, the limit of detection in our study was 1.2–4.8 viral RNA copies in comparison with 5.9–7.2 from previous study.
Of greatest interest, this new duplex assay improved the detection ability toward GI, being comparable to that for GII, based on the theoretical detection limits represented by the y-intercepts (44.26 for GI and 42.96 for GII), unlike that described in other studies, where a more sensitive detection was observed for GII [28, 30, 31]. This indicated a successful optimization of the duplex conditions by introducing the MGB quencher for the GI probe. When the qRT-PCR assay was further evaluated using RNA templates from rotavirus, adenovirus, and poliovirus, no cross-reactivity was observed in either duplex or simplex formats (data not shown).
Comparison between the duplex qRT-PCR and cRT-PCR assays
To examine the performance of the duplex qRT-PCR assay, we carried out the duplex assay against the 82 stool samples and 60 concentrated water samples. Based on the Kojima and Boxman cRT-PCR assays and our duplex qRT-PCR assay, we found that clinical (n = 76) samples were positive for NoV: GI (n = 2), GII (n = 64), and mixed GI and GII (n = 10), whereas the environmental (n = 25) samples showed NoV as GI (n = 5), GII (n = 14), and mixed GI and GII (n = 6) (data not shown). As a result, we identified a total of 23 GI and 94 GII NoV isolates in our tested specimens, and most of these (n = 82) were genotyped as listed in Table 1 and Fig. 1. Interestingly, the environmental samples displayed a significantly higher rate of GI detection when compared with the clinical samples (P = 0.0097).
As summarized in the Venn diagram displayed in Fig. 2, the detection capability of the duplex assay was much higher than that of both cRT-PCR assays. Our duplex assay detected 21 GI and 91 GII (i.e., 112/117); only five isolates (two clinical and two environmental) undetected by our assay were detected by Kojima assay (three clinical and one environmental) or by Boxman assay (one environmental), whose detection rates were 80/117 and 55/117, respectively. As shown in Table 4, it is noticeable that the duplex assay detected both GI and GII from the samples with mixed GI and GII (detection rate of 14/16), in contrast to the detection rates by the cRT-PCR assays, where the Boxman assay showed better detection ability than the Kojima assay (4/16 vs. 1/16). Moreover, the reason why we were able to check even low copy numbers in realtime qRT-PCR was that a high y-intercept value was obtained. As all the sample amplification results were included within this y-intercept value on equation in this study, its copy number value of all test samples was acceptable.

Number of positive samples detected by duplex qRT-PCR and cRT-PCR assays. Venn diagrams for GI (left) and GII (right) showing the number of samples positively detected by our duplex assay (black), cRT-PCR assay by Kojima (blue), and/or cRT-PCR assay by Boxman (red) against 82 clinical (upper) and 60 environmental (lower) specimens. The combination of these comparisons by Venn diagrams illustrates the number of samples commonly detected by all three assays and samples undetected by one or two of the assays. The number of samples from which NoV detection failed by any of the assays is not designated (Color figure online)
Discussion
NoV is the most common cause of nonbacterial acute gastroenteritis, having caused numerous recent outbreaks worldwide, with a broad genetic diversity that constitutes largely two genogroups (GI and GII) [32]. Fourteen genotypes of GI (GI-1 to GI-14) and 17 genotypes of GII (GII-1 to GII-17) contain most of the strains from clinical and environmental reservoirs [8]. The emerging significance of NoV outbreaks and the extensive diversity between and within the NoV genogroups require a more efficient (i.e., rapid, accurate/specific, sensitive, selective, comprehensive, and cost-effective) detection method for this viral species. In this study, we developed and evaluated a new TaqMan-based duplex qRT-PCR method to broadly and simultaneously detect, as well as to discriminate between, GI and GII genogroups of NoV. The assay was validated using NoV isolates from both clinical and environmental specimens collected in South Korea and comparing the results with those of two cRT-PCR assays using primer sets designed by Kojima et al. [22] and Boxman et al. [23].
There is a high degree of sequence variation among NoV isolates, and these variations are relatively scattered across the genome [33]. Thus, it is practically difficult to detect NoVs comprehensively due to the challenge of designing appropriate primer sets for PCR-based detection. In the case of GI and GII NoV isolates, however, short (<100 bp) stretches of the most strongly conserved nucleotides are found at the junction region between ORF1 and ORF2 in both GI and GII [18], which were also observed in our tested NoV isolates. Because the strongly conserved region is relatively short, we proposed that the TaqMan-based real-time detection system may be more effective for the broad detection of NoV isolates worldwide compared with the conventional gel electrophoresis-based detection systems used in cRT-PCR, with regards to time, reliability, and sensitivity.
The primer sets that had been designed previously for the sensitive and broadly reactive detection of GI and GII NoV from stool specimens or contaminated shellfish samples were modified slightly to optimize the duplex format [7, 28]. To ensure the simultaneous detection of GI and GII without the loss of efficiency, as in the simplex format, we introduced MGB to the GI probe to compensate for the hybridization instability by the shorter sequences of perfect match in the region of GI isolates, compared with that of GII isolates, as described previously [19, 20]. We were able to minimize degenerate nucleotides and replace inosine with two mixed pyrimidine nucleotides in the GI forward primer.
Because our assay exploits the TaqMan system, we achieved the quantitative detection of NoV isolates. A relatively broad linearity was observed in the duplex format by the use of serially diluted standard RNA transcripts (1–1010 copies per 25 μl for both GI and GII), which were comparable to the cognate values in the simplex formats under our conditions and those described by others [7, 28]. These results indicated that our developed assay could be used for the detection and quantification of NoV in both clinical and environmental specimens.
Recently, Nordgren et al. [31] reported a LUX real-time PCR assay for quantitative NoV detection from clinical specimens and for the discrimination between the two genogroups by first-derivative dissociation curves based on the difference between the melting properties of GI and GII amplicons. As in the previous TaqMan-based duplex assay [28], we were able to detect simultaneously both GI and GII more rapidly in an one-step reaction, without the loss of sensitivity, selectivity, and reliability. The sensitivity and efficiency could appear to be slightly better in our assay than in the assay by Jothikumar et al. [28] based on the slope and the y-intercept, respectively. More importantly, under their conditions, the theoretical sensitivity (reflected by the y-intercepts) was lower in the duplex format, whereas our assay did not significantly affect both the slopes and y-intercepts obtained using the simplex format. These differences are likely due to the selection of primers and probe sets and/or to the amplification conditions, which were better optimized in our duplex assay. The exploitation of BHQ in the GI probe could balance the slight difference between GI and GII properties and the annealing–extension combination at 60°C may minimize the ramp time and the temperature variation during the segment change in PCR amplification.
Although this assay was more successful for the detection of a broad range of NoV isolates from Korean samples, we still observed five NoV (two GI and three GII) isolates that were undetectable by our method. This kind of inferiority of the qRT-PCR assay compared with cRT-PCR is not without precedent. Jothikumar et al. [28] reported three cases that were undetectable by TaqMan assay, but were detected by cRT-PCR using nested primers designed by Häfliger et al. [34], which might be the same as those in the Boxman assay exploited in our study. Nonetheless, we had only one case in GI and none in GII that were detectable by Boxman assay. This implies that they might have identified more positive cases by using more than one cRT-PCR assay as in this study. In this regard, our duplex assay represents a notable improvement over the previous assay. Further improvement of our duplex assay is still underway to detect the previously undetectable samples by slightly modifying the protocols for sample extraction and/or RNA isolation, although these have been relatively standardized in many detection methods.
GI-1, GII-1, 3, and 4 types were detected in the environment samples used in this study. The study which was performed to detect viruses from river water in Korea, which has been already reported, also revealed that GI-1 was one of the major genotypes [35]. This study also reported the same result. It can be assumed that the highly prevalent human genotypes might have immigrated from human feces to the environment before being discovered [36]. Besides, the fact that more various genotypes were found in clinical samples than in environment samples may prove that genetic variation of this virus is high. In other words, these results show it is likely that, because of the characteristics of this virus, variation may occur through the amplification in a host. And when these epidemiological investigations will be performed periodically, the trend of genetic and immunological variation of this virus can be continuously monitored [15, 16].
Jothikumar et al. [28] have reported the reliable use of TaqMan technology for the simultaneous and genogroup-specific detection of NoV from stool specimens and naturally contaminated food samples. To the best of our knowledge, the newly developed TaqMan-based duplex real-time assay is very sensitive and specific for the simultaneous and quantitative genogroup-specific detection of prevalent NoV GI and GII isolates from both clinical and environmental specimens. Based on this assay, we were able to detect NoV isolates that were undetectable by other cRT-PCR assays. More importantly, this assay has been shown to display no cross-reactivity toward rotavirus, adenovirus, and poliovirus, demonstrating the NoV-specific detection in gastroenteritis cases. Our assay may be applicable to the rapid detection of NoV in foods, as well as to the monitoring of water reservoirs such as sewage and surface water. One of limitations of this study is that our assay was not designed nor tested for NoV genogroup IV, which is known to infect human as well. It should be designed and evaluated in future. Regardless, we believe that this assay method will have a tremendous impact on the routine diagnosis and epidemiological elucidation of NoV infections, leading and leads to a better maneuver in the public health control of this viral gastroenteritis in the future.
References
D.J. Allen, J.J. Gray, C.I. Gallimore, J. Xerry, M. Iturriza-Gomara, PLoS One 3, e1485 (2008)
T. Ando, J.S. Noel, R.L. Fankhauser, J. Infect. Dis. 181(Suppl. 2), S336–S348 (2000)
R.L. Fankhauser, S.S. Monroe, J.S. Noel, C.D. Humphrey, J.S. Bresee, U.D. Parashar, T. Ando, R.I. Glass, J. Infect. Dis. 186, 1–7 (2002)
A.M. Hutson, R.L. Atmar, M.K. Estes, Trends Microbiol. 12, 279–287 (2004)
M.M. Patel, A.J. Hall, J. Vinje, U.D. Parashar, J. Clin. Virol. 44, 1–8 (2009)
A. Leiste, A. Skaletz-Rorowski, I. Venten, P. Altmeyer, N.H. Brockmeyer, J. Dtsch. Dermatol. Ges. 6, 563–565 (2008)
T. Kageyama, S. Kojima, M. Shinohara, K. Uchida, S. Fukushi, F.B. Hoshino, N. Takeda, K. Katayama, J. Clin. Microbiol. 41, 1548–1557 (2003)
T. Kageyama, M. Shinogara, K. Uchida, S. Fukushi, F.B. Hoshino, S. Kojima, R. Takai, T. Oka, N. Takeda, K. Katayama, J. Clin. Microbiol. 42, 2988–2995 (2004)
F. Loisy, R.L. Atmar, P. Guillon, P. Le Cann, M. Pommepuy, F.S. Le Guyader, J. Virol. Methods 123(1), 1–7 (2005)
A.K. Da Silva, J.C. Le Saux, S. Parnaudeau, M. Pommepuy, M. Elimelech, F.S. Le Guyader, Appl. Environ. Microbiol. 73(24), 7891–7897 (2007)
S. Butot, F.S. Le Guyader, J. Krol, T. Putallaz, R. Amoroso, G. Sánchez, J. Virol. Methods 167(1), 90–94 (2010)
R.L. Atmar, M.K. Estes, Clin. Microbiol. Rev. 14, 15–37 (2001)
R.I. Glass, J. Noel, T. Ando, R. Fankhauser, G. Belliot, A. Mounts, U.D. Parashar, J.S. Bresee, S.S. Monroe, J. Infect. Dis. 181(Suppl. 2), S254–S261 (2000)
J.J. Siebenga, M.F. Beersma, H. Vennema, P. Van Biezen, N.J. Hartwig, M. Koopmans, J. Infect. Dis. 198, 994–1001 (2008)
C.I. Gallimore, M. Iturriza-Gomara, J. Xerry, J. Adigwe, J.J. Gray, Arch. Virol. 152, 1295–1303 (2007)
B. Lopman, H. Vennema, E. Kohli, P. Pothier, A. Sanchez, A. Negredo, J. Buesa, E. Schreier, M. Reacher, D. Brown, J. Gray, M. Iturriza, C. Gallimore, B. Bottiger, K.O. Hedlund, M. Torven, C.H. Von Bonsdorff, L. Maunula, M. Poljsak-Prijatelj, J. Zimsek, G. Reuter, G. Szucs, B. Melegh, L. Svennson, Y. Van Duijnhoven, M. Koopmans, Lancet 363, 682–688 (2004)
J. Xerry, C.I. Gallimore, M. Iturriza-Gomara, D.J. Allen, J.J. Gray, J. Clin. Microbiol. 46, 947–953 (2008)
R.A. Bull, G.S. Hansman, L.E. Clancy, M.M. Tanaka, W.D. Rawlinson, P.A. White, Emerg. Infect. Dis. 11, 1079–1085 (2005)
K. Bok, E.J. Abente, M. Realpe-Quintero, T. Mitra, S.V. Sosnovtsev, A.Z. Kapikian, K.Y. Green, J. Virol. 83, 11890–11901 (2009)
M. Hoehne, E. Schreier, BMC Infect. Dis. 6, 69 (2006)
H. Lee, M. Kim, S.Y. Paik, C.H. Lee, W.H. Jheong, J. Kim, G. Ko, J. Water Health 9(1), 27–36 (2011)
S. Kojima, T. Kageyama, S. Fukushi, F.B. Hoshino, M. Shinohara, K. Uchida, K. Natori, N. Takeda, K. Katayama, J. Virol, Methods 100, 107–114 (2002)
I.L. Boxman, J.J. Tilburg, T.A. Te Loeke, H. Vennema, K. Jonker, E. De Boer, M. Koopmans, Int. J. Food Microbiol. 108, 391–396 (2006)
Y. Park, Y.H. Cho, Y. Jee, G. Ko, Appl. Environ. Microbiol. 74, 4226–4230 (2008)
R. Rasmussen, Quantification on the LightCycler, in Rapid Cycle Real-time PCR, Methods and Applications, ed. by S. Meuer, C. Wittwer, K.-I. Nakagawara (Springer, Heidelberg, 2001), pp. 21–34
S.H. Kim, D.S. Cheon, J.H. Kim, D.H. Lee, W.H. Jheong, Y.J. Heo, H.M. Chung, Y. Jee, J.S. Lee, J. Clin. Microbiol. 43, 4836–4839 (2005)
T.G. Phan, K. Kaneshi, Y. Ueda, S. Nakaya, S. Nishimura, A. Yamamoto, K. Sugita, S. Takanashi, S. Okitsu, H. Ushijima, J. Med. Virol. 79, 1388–1400 (2007)
N. Jothikumar, J.A. Lowther, K. Henshilwood, D.N. Lees, V.R. Hill, J. Vinje, Appl. Environ. Microbiol. 71, 1870–1875 (2005)
I.V. Kutyavin, I.A. Afonina, A. Mills, V.V. Gorn, E.A. Lukhtanov, E.S. Belousov, M.J. Singer, D.K. Walburger, S.G. Lokhov, A.A. Gall, R. Dempcy, M.W. Reed, R.B. Meyer, J. Hedgpeth, Nucleic Acids Res. 28, 655–661 (2000)
R. Morales-Rayas, P.F.G. Wolffs, M.W. Griffiths, Int. J. Food Microbiol. 139, 48–55 (2010)
J. Nordgren, F. Bucardo, O. Dienus, L. Svensson, P.E. Lindgren, J. Clin. Microbiol. 46, 164–170 (2008)
G.S. Hansman, K. Natori, H. Shirato-horikoshi, S. Ogawa, T. Oka, K. Ktayama, T. Tanaka, T. Miyoshi, K. Sakae, S. Kobayashi, M. Shinohara, K. Uchida, N. Sakurai, K. Shinozaki, M. Okada, Y. Seto, K. Kamata, N. Nagata, K. Tanaka, T. Miyamura, N. Takeda, J. Gen. Virol. 87, 909–919 (2006)
J.J. Siebenga, H. Vennema, D.P. Zheng, J. Vinje, B.E. Lee, X.L. Pang, E.C. Ho, W. Lim, A. Choudekar, S. Broor, T. Halperin, N.B. Rasool, J. Hewitt, G.E. Greening, M. Jin, Z.J. Duan, Y. Lucero, M. O’Ryan, M. Hoehne, E. Schreier, R.M. Ratcliff, P.A. White, N. Iritani, G. Reuter, M. Koopmans, J. Infect. Dis. 200, 802–812 (2009)
D. Häfliger, M. Gilgen, J. Luthy, P. Hubner, Int. J. Food Microbiol. 37, 27–36 (1997)
C. Lee, S.J. Kim, Water Res. 42(17), 4477–4484 (2008)
D.J. Allen, J.J. Gray, C.I. Gallimore, J. Xerry, M. Iturriza-Gómara, PLoS One 3(1), e1485 (2008)
S. Ishida, S. Yoshizumi, T. Ikeda, M. Miyoshi, M. Okano, T. Okui, J. Med. Virol. 80, 913–920 (2008)
Acknowledgment
This research was supported by the Agriculture Research Center program of the Ministry of Food, Agriculture, Forestry, and Fisheries, and National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (2011-0000177).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Park, Y., Cho, YH. & Ko, G. A duplex real-time RT-PCR assay for the simultaneous genogroup-specific detection of noroviruses in both clinical and environmental specimens. Virus Genes 43, 192–200 (2011). https://doi.org/10.1007/s11262-011-0626-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-011-0626-4