
Abstract
Puumala virus (PUUV) is the predominant hantavirus species in Germany causing large numbers of mild to moderate cases of haemorrhagic fever with renal syndrome (HFRS). During an outbreak in South-East Germany in 2004 a novel PUUV subtype designated Bavaria was identified as the causative agent of HFRS in humans [1]. Here we present a molecular characterization of this PUUV strain by investigating novel partial and almost entire nucleocapsid (N) protein-encoding small (S-) segment sequences and partial medium (M-) segment sequences from bank voles (Myodes glareolus) trapped in Lower Bavaria during 2004 and 2005. Phylogenetic analyses confirmed their classification as subtype Bavaria, which is further subdivided into four geographical clusters. The entire N protein, harbouring an amino-terminal hexahistidine tag, of the Bavarian strain was produced in yeast Saccharomyces cerevisiae and showed a slightly different reactivity with N-specific monoclonal antibodies, compared to the yeast-expressed N protein of the PUUV strain Vranica/Hällnäs. Endpoint titration of human sera from different parts of Germany and from Finland revealed only very slight differences in the diagnostic value of the different recombinant proteins. Based on the novel N antigen indirect and monoclonal antibody capture IgG-ELISAs were established. By using serum panels from Germany and Finland their validation demonstrated a high sensitivity and specificity. In summary, our investigations demonstrated the Bavarian PUUV strain to be genetically divergent from other PUUV strains and the potential of its N protein for diagnostic applications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hantaviruses (family Bunyaviridae) are enveloped viruses with a segmented RNA genome of negative polarity [2]. The spherical particles of about 80–120 nm in diameter contain the three genome segments, small (S), medium (M) and large (L), which are complexed with the viral nucleocapsid (N) protein and associated at their ends with the RNA-dependent RNA polymerase [3]. The S-segment (1.7–2.1 kilo bases, kb) encodes the N protein. In addition, for Puumala virus (PUUV) and related hantaviruses a second open reading frame (ORF) was observed encoding a putative non-structural protein that was suggested to play a role in pathogenicity in humans or alternatively in the adaptation of the virus to the rodent host [4–6]. The M-segment (3.6–3.7 kb) encodes for a glycoprotein precursor that is co-translationally cleaved into the glycoproteins (Gn) G1 and Gc (G2). The L-segment (6.5–6.6 kb) encodes the RNA-dependent RNA polymerase [3].
Hantaviruses are carried and transmitted to humans by persistently infected rodents which shed the virus by urine, faeces and saliva. The major route of transmission to humans is by inhalation of virus-contaminated aerosols. Further, rodent biting is a rare transmission route to humans [7]. Human-to-human transmission has been described exclusively for the South-American Andes virus (ANDV; [8, 9]). In addition to rodent-associated hantaviruses there is an increasing number of newly detected hantaviruses in different insectivores [10–15]). However, it is unknown if these viruses can be transmitted to humans or cause disease.
In Europe, the bank vole (Myodes glareolus) associated PUUV is the geographically most widely distributed human pathogenic hantavirus. In addition, PUUV-like viruses were detected in M. rufocanus and M. regulus in Asia [16–18]. PUUV infections in humans may induce nephropathia epidemica (NE) which is characterized as a mild to moderate form of haemorrhagic fever with renal syndrome (HFRS) with a case fatality rate of less than 1% [19]. Reflecting the distribution of the reservoir host in almost all parts of Europe, human PUUV infections have been observed in different countries of northern, eastern, central, western and southern Europe [20–23]. In Germany, most NE cases are due to autochthonous PUUV infections. This is reflected in results of large-scale seroepidemiological studies as well as in the number of NE cases recorded since the introduction of the German Federal Protection against Infection Act in 2001 [24, 25]. The increased number of cases observed in 2005 (447 cases), in 2007 (1,688 cases) and in 2010 (2,016 cases) were caused by PUUV outbreaks mainly affecting Baden-Wuerttemberg, Bavaria, North Rhine Westphalia and Lower Saxony (Robert Koch-Institut: SurvStat, http://www3.rki.de/SurvStat, 16th March 2011; [26–28]). During an outbreak of human PUUV infections in Lower Bavaria in 2004, studies on rodents as well as on patients revealed a novel PUUV subtype designated Bavaria (Bayerischer Wald, Bawa, PUUV-Bawa; [1, 29]).
Human hantavirus infections are mainly detected by serological assays. Indirect immunofluorescence assays based on hantavirus-infected Vero cells, ELISA, immunoblot and immunochromatographical rapid tests have been developed. Mostly N protein derivatives of different hantavirus species generated by heterologous expression in Escherichia coli, yeast Saccharomyces cerevisiae and baculovirus-mediated insect cell systems are used as diagnostic antigens [30–32]. Although the N proteins of hantaviruses, in particular of closely related species, are cross-reactive, the use of homologous antigens of local virus strains was recommended for a sensitive detection of specific antibodies, especially during the acute phase [33–38].
Here we report on the phylogenetic characterization of the almost entire N protein-encoding sequence and non-coding regions (NCR) of the S-segment and partial M-segment sequences of PUUV-Bawa. The N protein of PUUV-Bawa was synthesized in yeast, immunologically characterized and tested for its diagnostic application in indirect and capture immunoglobulin G (IgG)-ELISA formats.
Materials and methods
Rodent trapping and necropsy
Rodents were collected in May 2005 at five locations in the administrative districts of Straubing-Bogen (military training grounds Bogen), Regen (military training grounds Regen; Hangenleiten—county Kirchberg) and Freyung-Grafenau (military training grounds Freyung; Raimundsreuth and Bierhütte—county Hohenau) in South-East Germany (Fig. 1; Table 1). Trapping, necropsy and analysis of rodents during the outbreak there in 2004 has been published before [1]. Blood and lung samples were taken from 19 animals (11 M. glareolus; four yellow-necked field mice Apodemus flavicollis; two long-tailed field mice A. sylvaticus; one common vole Microtus arvalis; one water vole Arvicola amphibius; see Table 1).

a Geographical map of Germany with the study area in the Federal State of Bavaria in the administrative districts Straubing-Bogen (B), Regen (R) and Freyung-Grafenau (F). b Administrative districts of Regen and Freyung-Grafenau with counties where PUUV-reactive rodents have been found are designated (2004: Lindberg (L), Kirchberg (K), Schöfweg (S), Hohenau (H), Freyung (F); 2005: Hohenau (H), Freyung (F), see also Table 1)
Human serum samples
For the establishment and the validation of the PUUV-Bawa antigen-based indirect and capture IgG-ELISAs and for cross-reactivity investigations, serum panels from Germany and Finland were applied. These sera have previously been analyzed by commercial reference assays and used for validation of indirect PUUV strain Vranica/Hällnäs (PUUV-Vra) IgM- and IgG-ELISAs ([32] and our unpublished data). The serum samples from Germany include a negative control panel of 149 sera from the routine diagnostics at the University Hospital in Frankfurt/Main and as positive control 79 sera from human NE cases. Out of these 79 sera, 51 samples were from PUUV-IgM- and PUUV-IgG-positive individuals with an acute infection and 28 from PUUV-IgM-negative and PUUV-IgG-positive persons at the convalescent phase. The set from Finland consists of 40 PUUV-IgM/PUUV-IgG-negative, 31 PUUV-IgG-positive and 40 PUUV-IgG/PUUV-IgM-positive sera.
Monoclonal antibodies
For antigenic comparison of the recombinant N proteins of PUUV-Bawa and PUUV-Vra monoclonal antibodies (mAbs) raised against N proteins of PUUV (mAbs 5E11, 5C5, 7A5, 2C6 [39]; 1C12, 5E1, 5A3, 2E12, 4C3 [40, 41]; A1C5 [42]), Hantaan virus (HTNV; E5/G6, Eco2 [43]; B5D9 [42]), Andes virus (ANDV; mAb 5C2/E10), Sin Nombre virus (SNV; mAb 5F1/F7; Immunological and Biochemical Testsystems GmbH, Reutlingen, Germany) and ANDV/SNV (4H3, 7G2 [44]) were used. The mAbs A1C5 and B5D9 were purchased from Progen Biotechnik GmbH (Heidelberg, Germany). Histidin (His)-tag specific mAbs 6E8 and 7B8 were obtained by immunization with a His-tagged yeast-expressed PUUV-Vra N protein (Zvirbliene et al. unpublished data).
RT-PCR and sequencing
Nucleic acids were extracted from M. glareolus lung and heart homogenates using the RNeasy kit (Qiagen, Hilden, Germany, see [1]). To obtain the NCRs as well as the entire N-encoding regions, several primer pairs (Table 2) were used for RT-PCR [see 1]. Partial M-segment sequences (nucleotides, nt 2369–3031, encoding a partial G2 of PUUV strain CG1820, accession number M29979) were amplified with primers C1 and C2 (Table 2) and the Superscript™ III RT/Platinum Taq Mix (Invitrogen, Karlsruhe, Germany) in a final volume of 50 μl according the manufacturer’s instructions. Following reverse transcription at 50°C for 30 min and denaturation at 94°C for 2 min, DNA was amplified in 55 cycles for 30 s at 94°C, 30 s at 50°C, 1 min at 68°C with a final extension for 5 min at 68°C. Morphological species determination of the PUUV-RT-PCR-positive M. glareolus was confirmed by PCR using mitochondrial cytochrome b (cyt b) gene specific primers as described before [1]. Direct sequencing was performed using segment and cyt b gene specific primers.
Cloning of the entire nucleocapsid protein-encoding sequence for expression in yeast
RNA extracted from homogenized lung tissue of the M. glareolus sample Bawa H-34/04 (Tables 1, 3; [1]) was amplified using primers PUUVexprF and PUUVexprR (Table 2) and the above described RT-PCR reagents with addition of 1 μM MgSO4. Cycling conditions were modified to 50 cycles for 25 s at 94°C, 50 s at 54°C, 75 s at 68°C. RT-PCR products of expected size were cloned into vector pCR2.1-TOPO® (TopoTA cloning kit, Invitrogen) as described by the manufacturer. The insert with a correct sequence encoding amino acids (aa) 2–433 of the PUUV-Bawa N protein was isolated as a XbaI fragment and subsequently inserted into XbaI-linearized pFX7-His6 plasmid [46]. The generation of the pFX7 expression plasmid encoding a His-tagged entire N protein of PUUV-Vra has been described previously [47].
Phylogenetic analysis
Nt sequences were aligned at the codon level using the ClustalW algorithm [48] implemented in BioEdit 7.0 [49] and revised manually. Phylogenetic relationships were inferred analogous to Braaker and Heckel [50] by neighbour-joining algorithms (NJ; [51]) implemented in Mega 3.1 [52] and by Bayesian algorithms (BI) implemented in MrBayes 3.1.2 [53]. The optimal mutation model was selected based on the Bayesian Information Criterion (BIC; [54]) in jModeltest 0.1.1 [55]: the TIM2ef model was best for the S-segment, the SYM + G model for the M-segment and the Tim1ef + I + G model for cyt b sequences. The NJ analyses were replicated 10,000 times using the Tamura-3-parameter model for the S-segment and cyt b data and the Maximum Composite Likelihood model [56] for the M-segment. The Bayesian analyses for the S- and M-segment were run three times for 2 million generations with every 10th generation sampled, using one cold and three heated chains. Bayesian cyt b data analyses were run for 10 million generations with every 100th generation samples with the web-based cluster implemented in the Cipres Portal [57]. The first 25% of the samples were discarded as burn-in and convergence was determined by examining the log likelihood values, the split frequencies and by using the web-based program AWTY [58]. Net average divergence based on the S- and M-segment was computed with Mega 3.1, using the mentioned nt substitution models. GenBank accession numbers of the sequences included in this study are shown in the corresponding phylogenetic trees.
Yeast synthesis and purification of the PUUV N antigens
The production in yeast cells and purification of N proteins of PUUV-Vra and PUUV-Bawa were done as described before [47]. Briefly, yeast S. cerevisiae cells of the wild-type strain FH4C were transformed with the corresponding pFX7-His6-derived yeast expression plasmids. After cultivation of the transformed yeast cells in YEPD growth medium (yeast extract 1%, peptone 2%, glucose 2%) supplemented with formaldehyde for 24 h at 28°C, N protein synthesis was induced by adding YEPG medium (yeast extract 1%, peptone 2%, galactose 6%) and incubation for additional 20 h. Protein purification was carried out by nickel-chelate affinity chromatography (Qiagen) with elution under denaturing conditions (8 M urea, 0.1 M NaH2PO4, 0.01 M Tris, pH 4.5; see [47]). For use in mAb-capture ELISA, the PUUV-Bawa N antigen was dialyzed against carbonate buffer as previously described [47].
Indirect PUUV-IgG-ELISA
The cross-reactivity of human sera with yeast-expressed N antigens of PUUV-Bawa and PUUV-Vra was investigated by endpoint titration in twofold steps starting at a dilution of 1:400 and using recently validated indirect IgG-ELISA protocols [32]. Definition of lower and upper cut-off values and determination of sensitivity and specificity for the PUUV-Bawa antigen-based ELISA follow published criteria [32].
PUUV-Bawa antigen-based IgG capture ELISA
For the capture ELISA Maxi-Sorp plates (Nunc) were coated with 100 μl/well capture mAb A1C5 (1:1,000 in carbonate buffer, pH 9.8) for 30 min at 37°C. After washing three times with PBS/0.1% Tween 20, blocking was performed using 200 μl/well 3% BSA in PBS/0.05% Tween 20 for 30 min at room temperature (RT). Additional washing was followed by adding 100 μl/well recombinant PUUV-Bawa N antigen (2 μg/ml in 1% BSA/0.05% Tween 20 in PBS) and incubation at 37°C for 30 min. Plates were washed and the human sera were added (diluted 1:400 in 1% BSA/0.05% Tween 20 in PBS). After incubation at 37°C for 30 min the plates were washed, the HRP-labelled goat anti-human IgG conjugate (1:2,000, Sigma-Aldrich, Deisenhofen, Germany) was incubated at 37°C for 1 h. The final steps of this ELISA as well as the definition of lower and upper cut-off values and the determination of sensitivity and specificity were done the same way as for the indirect ELISA.
SDS-PAGE and western blot analysis
Sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analyses using an enhanced chemiluminescence (ECL) reagent (Amersham, Braunschweig, Germany) were performed as previously described [32, 37]. Briefly, 2.5 μg of PUUV-Vra or PUUV-Bawa N proteins, each in a volume of 250 μl, were separated in a 12.5% SDS-PAGE gel with a 0.75 mm comb in a Mini-Protean 3 system (BioRad, Munich, Germany). Thereafter, the proteins were semidry blotted onto a Polyvinylidenfluoride (PVDF) membrane (Millipore, Bedford, MA). After blocking, 3 mm-wide strips of the membrane were incubated for 90 min at RT with 500 μl mAb diluted 1:500 in blocking buffer. For detecting the immune reaction, the strips were incubated for 90 min at RT in 500 μl HRP-labelled goat anti-mouse IgG (H + L) (BioRad) diluted 1:500 in blocking buffer.
Results and discussion
PUUV strain Bavaria is persistently present in M. glareolus in Lower Bavaria
In this study we describe the hantavirus screening of 19 rodents trapped in Lower Bavaria in 2005. Lung samples from 5 of 11 (45%) bank voles were found to be positive in at least one of the PUUV-specific RT-PCRs. All eight lung samples from other rodent species were negative (Table 1). This finding is in line with the prevalence observed in two independent studies in the same region during 2004. In a combined serological and RT-PCR study 10 of 29 (34.5%) of bank voles were found to be PUUV-positive [1]. A similar prevalence of 21% (8/38) was observed in a RT-PCR screening of bank voles from this particular area in November 2004 [29]. Interestingly, the current study demonstrated a continuing presence of PUUV-infected bank voles at least in the trapping area Hohenau-Raimundsreuth (Fig. 1; Table 1). In this area PUUV-infected bank voles were detected both in 2004 and 2005 suggesting an overwinter survival of at least some of the infected animals or, alternatively, a long-term viability of infectious virus particles in the environment. The latter might be supported by a relatively long-term stability of hantaviruses outside of their host [59, 60]. A further longitudinal study in South-East Germany during 2008–2010 has recently shown that the PUUV strain Bavaria is still present in the study area (see [61]; Essbauer et al. unpublished data).
Molecular and phylogenetic analysis of partial S- and M-segment sequences of PUUV strain Bavaria illustrate the high variability of PUUV in Germany
In this study, for two M. glareolus trapped in Lower Bavaria in 2005 (Bawa F-151/05, 1711 nt; Bawa F-159/05, 1672 nt) the almost entire PUUV S-segment sequences were amplified. For two samples (Bawa H-152/05, 1603 nt; Bawa F-157/05, 1639 nt) we obtained the main part of the N protein-encoding region and part of the 3′-NCR. For the lung sample of M. glareolus Bawa F-153/05 an amplification product was exclusively obtained using a nested RT-PCR (Table 3). These novel PUUV sequences are highly similar to the subtype Bawa sequences collected from M. glareolus in this region in 2004 [1, 29]. For seven out of ten animals trapped in 2004 [1], the S-segment sequences were extended up to about 1,700 nt (see Tables 1, 3). Finally, for 10 out of the 11 PUUV S-segment RT-PCR-positive M. glareolus trapped in 2004 and 2005, partial M-segment sequences were amplified from lung tissue (Table 3).
Phylogenetic analyses of 65 partial S-segment sequences (1,302 nt) demonstrated strong evolutionary differentiation of PUUV from the outgroup sequences (Fig. 2a). Furthermore, the analyses revealed a strong geographical clustering within PUUV into a Central European, Northern European I, Northern and Eastern European, Northern European II and Asian clade. The PUUV sequences from Bavaria (Southern Germany) were clearly separated from other German sequences (Western Germany I, Western Germany II) within the Central European cluster. Further, analyses of 32 M-segment sequences (621 nt) confirmed a clear separation of the PUUV from the outgroup sequences (Fig. 3a). As for the S-Segment, a well supported geographical grouping into a Southern German, Western German-Belgian, Swedish and Finish-Russian cluster was revealed, and the sequences from Bavaria (Southern Germany) were distinct from other sequences from Germany (Western Germany).

Bayesian reconstruction of phylogenetic relationships based on 1,302 nt S-segment sequences. Support values are displayed only for main nodes that connect major evolutionary lineages. Posterior probabilities are indicated above the major branches and percentage of bootstrap support for neighbour-joining (NJ) algorithms below the branches. *A different topology based on NJ algorithms, ns refers to posterior probabilities <0.70 and percentages of bootstrap support <70%. a Phylogenetic relationships of PUUV sequences with closely related viruses used as outgroup to root the tree. The new Bavarian cluster is highlighted in grey. b The Bavarian cluster displayed with the four trapping sites Hohenau, Lindberg, Kirchberg and Freyung. The abbreviation W1 indicates the mitochondrial affiliation to the Western European 1 cluster, W2 the association to the Western European 2 cluster (see Supplementary figure)

Bayesian reconstruction of phylogenetic relationships based on 621 nt M-segment sequences. Only support values for main nodes that connect major evolutionary lineages are displayed. Posterior probabilities are indicated above the major branches and percentage of bootstrap support for neighbour-joining (NJ) algorithms below the branches. *A different topology based on NJ algorithms, ns refers to posterior probabilities <0.70 and percentages of bootstrap support <70%. a Phylogenetic relationships of PUUV sequences with closely related viruses used as outgroup to root the tree. The new Bavarian cluster is highlighted in grey. b The Bavarian cluster is displayed with the four trapping sites Hohenau, Lindberg, Kirchberg and Freyung. The abbreviation W1 indicates the mitochondrial affiliation to the Western European 1 cluster, W2 the association to the Western European 2 cluster (see Supplementary figure)
The clear separation of the Bavarian clade from PUUV sequences from the western parts of Germany is reflected in nt divergences between German clades of 12.3 and 23.3% based on the S-segment and of 19.7% in the M-segment (Table 4a, b). Based on corresponding aa, the Bavarian cluster differed 0.6 and 1% from the Western Germany clusters for the S-segment and 1.5% for the M-segment. The other Central European clusters have a nt divergence in the partial S-segment between 20.5% (Belgium) and 30.3% (Austria) to the Bavarian strain, resulting in an aa divergence up to 1.8% (Table 4a). The majority of aa exchanges was found in the region between aa 247 and 265 of the N protein. The divergence of the Bavarian cluster to the other PUUV clusters in the M-segment ranged from 24.6 to 24.7% at the nt and 2–4.2% at the aa level (Table 4b).
Sequence variation of PUUV at small geographical scale and corresponding mitochondrial (mt) DNA sequence analysis of M. glareolus
A more detailed evaluation of the Bavarian S- and M-segment sequences showed a geographical clustering associated with the regions Hohenau, Lindberg, Kirchberg and Hohenau/Freyung (Figs. 1, 2b, 3b). For the almost entire S-segment and the entire N protein-encoding sequences, the difference between the sub-clusters within the Bavarian clade (Fig. 2b) ranged from 0.4 to 3.1% at the nt level, and from 0% up to 1% on the aa level. The partial 3′-NCR sequences (377 nt) of 11 samples revealed an intra-group nt divergence from 0 up to 12.3%. In comparison, the nt divergence of the partial M-segment among the four Bavarian sub-clusters (Fig. 3b) ranged from 0.9 to 3.2%, resulting in an aa divergence of 0–0.5%.
For other PUUV lineages we have so far only very limited data concerning divergence within one trapping site at a defined time point. For 17 sequences of the PUUV originating from the city of Cologne in 2005 a divergence of up to 1.2% for the 722 nt long partial S-segment was found [26, 62]. In comparison the genetic diversity of 23 PUUV sequences from Finland was comparable to the divergence described herein, i.e. 0.2–4.9% for the S- and 0.2–4.8% for the M-segment [63]. Similarly, a divergence of 0–2.6% for the S-, and 0–0.8% for the partial M-segment were reported for Russian PUUV strains [64]. This relatively low local differentiation among virus strains is similar to patterns detected in Tula virus where strain similarity was associated with trapping locality but not trapping year or vole host species [65]. It is noteworthy that PUUV similarity in the present study was associated with trapping locality even at distances between trapping sites of only about 50 km (Figs. 2b, 3b). This may suggest relatively independent virus evolution between sites at the regional scale and low or no direct migration among the host populations.
Voles carrying PUUV strain Bawa were all confirmed as M. glareolus by sequence analysis of approximately 968 nt of the cyt b gene. Phylogenetic analysis further revealed that M. glareolus in Bavaria clustered together with other sequences from a large region in Western Europe, which is evolutionarily distinct from other genetic lineages in the species occurring in Italy, the Balkans, Spain or Eastern Europe (Supplementary Figure; [64, 66, 67]). However, M. glareolus from Bavaria belong to 2 weakly defined different subclusters (W1 and W2) of the Western European lineage. The presence of haplotypes from these two sub-clusters within single populations is not unusual [66]. Given the relatively low geographic resolution of cyt b within lineages [66, 68, 69], it is not surprising that this marker failed to resolve potential differentiation among voles from the Bavarian sites. Evolutionary analyses of M. glareolus populations have shown that genetic differentiation and thus limited dispersal may exist at regional and local scales [67; Jenkins et al. unpublished]. However, the study of population connectivity and migration at such geographical scales requires typically the analysis of highly variable genetic markers and dedicated population genetics analyses of the virus hosts (see [70–72]).
Reactivity of human sera and mAbs with different PUUV N antigens
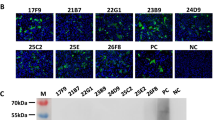
The recombinant N protein of PUUV-Bawa was shown to be highly pure by SDS-PAGE and to have the expected molecular weight of 49.4 kDa (data not shown). The high-level expression and purification of large quantities of this antigen highlighted the value of the used yeast expression system (see [46]). A western blot analysis of the protein with the His-tag specific mAbs 6E8 and 7B8 confirmed the presence of the amino-terminal His-tag on the recombinant N protein (Fig. 4).

Western blot reactivity of the purified recombinant PUUV-Bawa N protein (aa 2–433) of about 49 kDa with His-tag specific mAbs 6E8 and 7B8 and mAbs raised against N proteins of PUUV (mAbs 5E11, 5C5, 2C6 [39], 5A3, 4C3, 1C12, A1C5 [42], 5E1, 2E12, [40, 41], 7A5), Sin Nombre virus (SNV; mAb 5F1/F7), Andes virus (ANDV; mAb 5C2/E10), ANDV/SNV (mAb 4H3, 7G2 [44]) and Hantaan virus (HTNV; mAb E5/G6, Eco2 [43]; B5D9 [42]). The epitope localization of the PUUV-, HTNV and SNV/ANDV-reactive mAbs has been described in detail in [43, 78, 79]. Epitope specificity for mAbs 5C2/E10 and 5F1/F7 is given according to the information of the manufacturer (Immunological and Biochemical Testsystems GmbH, Reutlingen, Germany)
The antigenicity of the PUUV-Bawa N protein was characterized with mAbs raised against PUUV, HTNV, SNV and ANDV. In general, the reactivity of the N protein of PUUV-Bawa was identical to that of PUUV-Vra, PUUV strain Sotkamo (PUUV-Sot) and PUUV strain Kazan (PUUV-Kaz) for the majority of mAbs (Fig. 4; [39, 44, 73]). In contrast, the mAb 2C6 raised against the 120 amino-terminal aa of PUUV-Vra N protein and mapped to an epitope between aa 1 and 45 did not react with PUUV-Bawa N antigen as previously reported for N antigens of PUUV-Kaz and PUUV-Sot and confirmed the strain specificity of this mAb [74]. Interestingly, mAb 5A3 mapped to an epitope in the same region [73] reacted vice versa and was found to be non-reactive with PUUV-Vra N protein [73] but reactive with that of PUUV-Bawa as previously seen for PUUV-Sot and PUUV-Kaz [46]. Taken together, based on the reactivity pattern of these two N-specific mAbs, the N protein of the PUUV-Bawa subtype was antigenically closer related to those from Finnish PUUV-Sot and Russian PUUV-Kaz strains than to that of the Swedish PUUV-Vra strain.
In addition, the aa sequence differences between the N proteins of these four PUUV strains in the 45 amino-terminal aa residues underline an important role of aa residue 35 in the epitope integrity for mAbs 2C6 and 5A3 ([44, 73]; see Fig. 5). Interestingly, at aa position 35 an aspartic acid (D) residue was found in the N protein of the Bavaria strain, as in the majority of the PUUV strains. In comparison, PUUV strains Vranica/Hällnas, P360 and K27 contain at the corresponding position a tyrosine (Y) residue (Fig. 5). The importance of aa residue 35 was also observed in a recent study describing the reactivity patterns of cell culture passage-derived PUUV variants with a N protein-specific mAb [75].

Amino acid sequences spanning residues 1–40 of N proteins of German PUUV strains Bavaria (BAWA K-9/04, AY954722; BAWA H-33/04, DQ016430; BAWA H-34/04, AY954723; BAWA L-41/04, DQ016432; BAWA F-151/05, EU439968; BAWA F-152/05, EU439969; BAWA F-157/05, EU439971; BAWA F-159/05, EU439972; marked in bold) and Heidelberg (DQ094844, PUUV strain Heidelberg/hu) in comparison to corresponding PUUV N protein sequences from other European countries (U14137, strain Vranica-Hällnäs; Z84204, strain Puu/Kazan; Z69985, strain Puu/Virrat/25Cg/95; AJ223368, strain Puu/Eidsvoll/1124v; AJ223377, strain Puu/Solleftea/Cg6/95; AF442613, strain CG17/Baskiria-2001; AJ314599, strain Baltic/205Cg/00; X61035, strain Sotkamo; M32750, strain CG1820; L11347, strain P360; L08804, strain K27; AF367065, strain CG168). The amino acid residue 35, most likely responsible for distinguishing reaction with mAbs 2C6 and 5A3 is marked
Establishment, validation and comparison of a PUUV-Bawa N protein-based indirect IgG and capture ELISA
Purified, yeast-expressed viral antigens have broadly been used for diagnostics of virus infections in humans and animals [76–81]. We have recently described indirect and mAb-capture IgG-ELISAs for the detection of human infections with SNV, ANDV, PUUV, DOBV and HTNV [32, 36–38]. A corresponding ELISA using the PUUV-Bawa N protein was established here following a recently published protocol for a PUUV-Vra N protein-based IgG-ELISA [32]. When validating this novel in-house ELISA with 100 negative and 76 positive control sera from Germany one sample was identified as false negative (Table 5). Therefore, the diagnostic sensitivity and specificity of the novel ELISA was 99 and 100%, respectively.
The novel IgG-ELISA was compared to the corresponding PUUV-Vra N protein-based ELISA by endpoint titration of 68 sera from different regions in Germany and 10 sera from Finland. In general, almost all sera showed a very similar reactivity with both N antigens. Thus the endpoint titer of 30 sera from Germany and 7 sera from Finland were identical to both antigens. The endpoint titer to the PUUV-Bawa antigen was twofold higher for 34 sera from Germany and two sera from Finland. For three sera from Germany the endpoint titer to PUUV-Bawa was found to be fourfold higher than that to PUUV-Vra. Only one serum originating from south Germany was detected by the PUUV-Vra IgG-ELISA at the initial dilution, but not at all with the corresponding PUUV-Bawa test (data not shown). The similar reactivity of the N proteins of PUUV-Bawa and PUUV-Vra with the majority of N protein-specific mAbs and the results of the cross-reactivity investigations with human sera demonstrated a close antigenic similarity of these N proteins. Future large-scale serological studies will have to prove if local homologous PUUV antigens are required for a highly sensitive detection of hantavirus-specific antibodies, as discussed previously [33–38, 82].
The initial validation of the herein described mAb-capture PUUV-IgG-ELISA with the panel from Germany revealed 148 true negative, 74 true positive and four false negative sera. The evaluation of the sera originating from Finland revealed 62 true positive, 40 true negative, but 8 false negative sera (Table 5).
In line with data obtained for yeast-expressed HTNV antigen [83, 84], we found a very similar sensitivity and specificity of the PUUV-Bawa antigen-based indirect ELISA (99 and 100%) and mAb-capture ELISA (95 and 100%) for the serum panel from Germany. A comparison of the results of the validation of the indirect PUUV-Vra ELISA and the PUUV-Bawa mAb-capture ELISA for all investigated sera from Finland showed a lower sensitivity for the capture ELISA (89%) than for the indirect ELISA (100%), but a higher specificity (100 and 95%, respectively). The lower sensitivity of the PUUV-Bawa capture ELISA may be a result of the heterologous antigen used in this assay with Finish serum samples. Alternatively, also a competition of the mAb and the serum samples cannot be excluded. The high sensitivity and specificity might be due to the use of a purified yeast-expressed antigen preventing problems observed for E. coli-expressed antigens [85].
In conclusion, these investigations suggest a continuing prevalence of PUUV subtype Bavaria in M. glareolus from Lower Bavaria. The encoded N protein can be expressed at high-level in yeast S. cerevisiae and is suitable for diagnostic purposes in an indirect and capture IgG-ELISA for human sera from Germany. Future large-scale seroepidemiological studies in different European regions should consider if this antigen might be applied for diagnostic purposes in these regions as well.
References
S. Essbauer, J. Schmidt, F.J. Conraths, R. Friedrich, J. Koch, W. Hautmann, M. Pfeffer, R. Wölfel, J. Finke, G. Dobler, R. Ulrich, A new Puumala hantavirus subtype in rodents associated with an outbreak of Nephropathia epidemica in South-East Germany in 2004. Epidemiol. Infect. 134, 1333–1344 (2006)
S.T. Nichol, B.J. Beaty, R.M. Elliott, R. Goldbach, A. Plyusnin, C.S. Schmaljohn, R.B. Tesh, Family Bunyaviridae, in Virus Taxonomy, ed. by C.M. Fauquet, M.A. Mayo, J. Maniloff, U. Desselberger, L.A. Ball. 8th Report of the International Committee on Taxonomy of Viruses (Elsevier Academic Press, San Diego, CA, 2005), pp. 695–716
C.S. Schmaljohn, S.T. Nichol, Bunyaviridae, in Fields Virology, vol. 2, 5th edn., ed. by D.M. Knipe, P.M. Howley (Lippencott, Williams & Wilkins, Philadelphia, PA, 2007), pp. 1741–1789
A. Plyusnin, S.P. Morzunov, Virus evolution and genetic diversity of hantaviruses and their rodent hosts. Curr. Top. Microbiol. Immunol. 256, 47–75 (2001)
R. Ulrich, B. Hjelle, C. Pitra, D.H. Krüger, Emerging viruses: the case “Hantavirus”. Intervirology 45, 318–327 (2002)
K.M. Jääskeläinen, A. Plyusnina, Å. Lundkvist, A. Vaheri, A. Plyusnin, Tula hantavirus isolate with the full-length ORF for nonstructural protein NSs survives for more consequent passages in interferon-competent cells than the isolate having truncated NSs ORF. Virol. J. 5, 3 (2008)
G. Schönrich, A. Rang, N. Lütteke, M.J. Raftery, N. Charbonnel, R.G. Ulrich, Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 225, 163–189 (2008)
R.M. Wells, S. Sosa Estani, Z.E. Yadon, D. Enria, P. Padula, N. Pini, J.N. Mills, C.J. Peters, E.L. Segura, An unusual hantavirus outbreak in southern Argentina: person-to-person transmission? Hantavirus Pulmonary Syndrome Study Group for Patagonia. Emerg. Infect. Dis. 3, 171–174 (1997)
P.J. Padula, A. Edelstein, S.D. Miguel, N.M. Lopez, C.M. Rossi, R.D. Rabinovich, Hantavirus pulmonary syndrome outbreak in Argentina: molecular evidence for person-to-person transmission of Andes virus. Virology 241, 323–330 (1998)
D.E. Carey, R. Reuben, K.N. Panicker, R.E. Shope, R.M. Myers, Thottapalayam virus: a presumptive arbovirus isolated from a shrew in India. Indian J. Med. Res. 59, 1758–1760 (1971)
J.W. Song, S.H. Gu, S.N. Bennett, S. Arai, M. Puorger, M. Hilbe, R. Yanagihara, Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol. J. 4, 114 (2007)
J.W. Song, H.J. Kang, K.J. Song, T.T. Truong, S.N. Bennett, S. Arai, N.U. Truong, R. Yanagihara, Newfound hantavirus in Chinese mole shrew, Vietnam. Emerg. Infect. Dis. 13, 1784–1787 (2007)
B. Klempa, E. Fichet-Calvet, E. Lecompte, B. Auste, V. Aniskin, H. Meisel, P. Barriere, L. Koivogui, J. ter Meulen, D.H. Krüger, Novel hantavirus sequences in shrew, Guinea. Emerg. Infect. Dis. 13, 520–522 (2007)
S. Arai, J.W. Song, L. Sumibcay, S.N. Bennett, V.R. Nerurkar, C. Parmenter, J.A. Cook, T.L. Yates, R. Yanagihara, Hantavirus in northern short-tailed shrew, United States. Emerg. Infect. Dis. 13, 1420–1423 (2007)
H.J. Kang, S.N. Bennett, L. Sumibcay, S. Arai, A.G. Hope, G. Mocz, J.W. Song, J.A. Cook, R. Yanagihara, Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS One 4, 7 (2009)
H. Kariwa, S. Yoshizumi, J. Arikawa, K. Yoshimatsu, K. Takahashi, I. Takashima, N. Hashimoto, Evidence for the existence of Puumula-related virus among Clethrionomys rufocanus in Hokkaido. Japan. Am. J. Trop. Med. Hyg. 53, 222–227 (1995)
N.H. Daud, H. Kariwa, Y. Tanikawa, I. Nakamura, T. Seto, D. Miyashita, K. Yoshii, M. Nakauchi, K. Yoshimatsu, J. Arikawa, I. Takashima, Mode of infection of Hokkaido virus (Genus Hantavirus) among grey red-backed voles, Myodes rufocanus, in Hokkaido, Japan. Microbiol. Immunol. 51, 1081–1090 (2007)
A. Plyusnina, J. Laakkonen, J. Niemimaa, K. Nemirov, G. Muruyeva, B. Pohodiev, Å. Lundkvist, A. Vaheri, H. Henttonen, O. Vapalahti, A. Plyusnin, Genetic analysis of hantaviruses carried by Myodes and Microtus rodents in Buryatia. Virol. J. 5, 4 (2008)
J. Mustonen, O. Vapalahti, H. Henttonen, A. Pasternack, A. Vaheri, Epidemiology of hantavirus infections in Europe. Nephrol. Dial. Transplant. 13, 2729–2731 (1998)
H. Kallio-Kokko, N. Uzcategui, O. Vapalahti, O. Vaheri, Viral zoonoses in Europe. FEMS Microbiol. Rev. 29, 1051–1077 (2005)
S. Sandmann, H. Meisel, A. Razanskiene, A. Wolbert, B. Pohl, D.H. Krüger, K. Sasnauskas, R. Ulrich, Detection of human hantavirus infections in Lithuania. Infection 33(2), 66–72 (2005)
L. Fontana-Binard, D. Schultze, B.S. Rojanavisut, D.H. Krüger, G. Dollenmaier, G. Zanetti, P. Meylan, First case of nephropathia epidemica acquired in Switzerland. Rev. Med. Suisse. 4(163), 1572–1575 (2008)
S. Grygorczuk, S. Pancewicz, J. Zajkowska, M. Kondrusik, R. Swierzbińska, A. Moniuszko, W. Pawlak-Zalewska, Detection of anti-hantavirus antibodies in forest workers in the north-east of Poland. Przegl. Epidemiol. 62, 531–537 (2008)
L. Zöller, M. Faulde, H. Meisel, B. Ruh, P. Kimmig, U. Schelling, M. Zeier, P. Kulzer, C. Becker, M. Roggendorf, E.K.F. Bautz, D.H. Krüger, G. Darai, Seroprevalence of hantavirus antibodies in Germany as determined by a new recombinant enzyme immunoassay. Eur. J. Clin. Microbiol. Infect. Dis. 14, 305–313 (1995)
R. Ulrich, H. Meisel, M. Schütt, J. Schmidt, A. Kunz, B. Klempa, M. Niedrig, P. Kimmig, G. Pauli, D.H. Krüger, J. Koch, Verbreitung von Hantavirusinfektionen in Deutschland. Bundesgesundheitsbl. Gesundheitsforsch. Gesundheitsschutz 47, 661–670 (2004)
S.S. Essbauer, J. Schmidt-Chanasit, E.L. Madeja, W. Wegener, R. Friedrich, R. Petraityte, K. Sasnauskas, J. Jacob, J. Koch, G. Dobler, F.J. Conraths, M. Pfeffer, C. Pitra, R.G. Ulrich, Nephropathia epidemica outbreak in a metropolitan area, Germany. Emerg. Infect. Dis. 13, 1271–1273 (2007)
J. Koch, S.O. Brockmann, C. Winter, P. Kimmig, K. Stark, Significant increase of hantavirus infections in Germany since the beginning of 2007. Euro Surveill. 12, 5 (2007)
J. Hofmann, H. Meisel, B. Klempa, S.M. Vesenbeckh, R. Beck, D. Michel, J. Schmidt-Chanasit, R.G. Ulrich, S. Grund, G. Enders, D.H. Krüger, Molecular epidemiology of a large hantavirus outbreak in Germany, 2007. Emerg. Infect. Dis. 14, 850–852 (2008)
S. Schilling, P. Emmerich, B. Klempa, B. Auste, E. Schnaith, H. Schmitz, D.H. Krüger, S. Günther, H. Meisel, Hantavirus outbreak in Germany: limitations of routine serological diagnostics and clustering of virus sequences of human and rodent origin. J. Clin. Microbiol. 45, 3008–3014 (2007)
H.W. Lee, C. Calisher, C.S. Schmaljohn, Manual of Hemorrhagic Fever with Renal Syndrome and Hantavirus Pulmonary Syndrome (WHO Collaborating Center for Virus Reference and Research, Seoul, 1999)
D.H. Krüger, R. Ulrich, Å. Lundkvist, Hantavirus infections and their prevention. Microbes. Infect. 3, 1129–1144 (2001)
M. Mertens, R. Wölfel, K. Ullrich, K. Yoshimatsu, J. Blumhardt, I. Römer, J. Esser, J. Schmidt-Chanasit, M.H. Groschup, G. Dobler, S.S. Essbauer, R.G. Ulrich, Seroepidemiological study in a Puumala virus outbreak area in South-East Germany. Med. Mircobiol. Immunol. 198, 83–91 (2009)
H. Feldmann, A. Sanchez, S. Morzunov, C.F. Spiropoulou, P.E. Rollin, T.G. Ksiazek, C.J. Peters, S.T. Nichol, Utilization of autopsy RNA for the synthesis of the nucleocapsid antigen of a newly recognized virus associated with hantavirus pulmonary syndrome. Virus Res. 30, 351–367 (1993)
F. Elgh, M. Linderholm, G. Wadell, A. Tarnvik, P. Juto, Development of humoral cross-reactivity to the nucleocapsid protein of heterologous hantaviruses in nephropathia epidemica. FEMS Immunol. Med. Microbiol. 22, 309–315 (1998)
K.B. Sjolander, Å. Lundkvist, Dobrava virus infection: serological diagnosis and cross-reactions to other hantaviruses. J. Virol. Methods 80(2), 137–143 (1999)
J. Schmidt, H. Meisel, B. Hjelle, D.H. Krüger, R. Ulrich, Development and evaluation of serological assays for detection of human hantavirus infections caused by Sin Nombre virus. J. Clin. Virol. 33, 247–253 (2005)
J. Schmidt, H. Meisel, S.G. Capria, R. Petraityte, Å. Lundkvist, B. Hjelle, P.A. Vial, P. Padula, D.H. Krüger, R. Ulrich, Serological assays for the detection of human Andes hantavirus infections based on its yeast-expressed nucleocapsid protein. Intervirology 49, 173–184 (2006)
H. Meisel, A. Wolbert, A. Razanskiene, A. Marg, A. Kazaks, K. Sasnauskas, G. Pauli, R. Ulrich, D.H. Krüger, Development of novel immunoglobulin G (IgG), IgA and IgM enzyme immunoassays based on recombinant Puumala and Dobrava hantavirus nucleocapsid proteins. Clin. Vaccine Immunol. 13, 1349–1357 (2006)
A. Zvirbliene, L. Samonskyte, A. Gedvilaite, T. Voronkova, R. Ulrich, K. Sasnauskas, Generation of monoclonal antibodies of desired specificity using chimeric polyomavirus-derived virus-like particles. J. Immunol. Methods 311, 57–70 (2006)
Å. Lundkvist, A. Fatouros, B. Niklasson, Antigenic variation of European haemorrhagic fever with renal syndrome virus strains characterized using bank vole monoclonal antibodies. J. Gen. Virol. 72, 2097–2103 (1991)
Å. Lundkvist, B. Niklasson, Bank vole monoclonal antibodies against Puumala virus envelope glycoproteins: identification of epitopes involved in neutralization. Arch. Virol. 126, 93–105 (1992)
L.G. Zöller, S. Yang, P. Gött, E.K. Bautz, G. Darai, A novel μ-capture enzyme-linked immunosorbent assay based on recombinant proteins for sensitive and specific diagnosis of hemorrhagic fever with renal syndrome. J. Clin. Microbiol. 31, 1194–1199 (1993)
K. Yoshimatsu, Y.C. Yoo, R. Yoshida, C. Ishihara, I. Azuma, J. Arikawa, Protective immunity of Hantaan virus nucleocapsid and envelope protein studied using baculovirus-expressed proteins. Arch. Virol. 130, 365–376 (1993)
I. Kucinskaite-Kodze, R. Petraityte-Burneikiene, A. Zvirbliene, B. Hjelle, R.A. Medina, A. Gedvilaite, A. Razanskiene, J. Schmidt-Chanasit, M. Mertens, P. Padula, K. Sasnauskas, R.G. Ulrich, Characterization of monoclonal antibodies against hantavirus nucleocapsid protein and their use for immunohistochemistry on rodent and human samples. Arch. Virol. 156, 443–456 (2011)
A. Plyusnin, J. Hörling, M. Kanerva, J. Mustonen, Y. Cheng, J. Partanen, O. Vapalahti, S.K. Kukkonen, J. Niemimaa, H. Henttonen, B. Niklasson, Å. Lundkvist, A. Vaheri, Puumala hantavirus genome in patients with nephropathia epidemica: correlation of PCR positivity with HLA haplotype and link to viral sequences in local rodents. J. Clin. Microbiol. 35, 1090–1096 (1997)
A. Razanskiene, J. Schmidt, A. Geldmacher, A. Ritzi, M. Niedrig, Å. Lundkvist, D.H. Krüger, H. Meisel, K. Sasnauskas, R. Ulrich, High yields of stable and highly pure nucleocapsid proteins of different hantaviruses can be generated in the yeast Saccharomyces cerevisiae. J. Biotechnol. 111, 319–333 (2004)
A. Dargeviciute, K. Brus Sjölander, K. Sasnauskas, D.H. Krüger, H. Meisel, R. Ulrich, Å. Lundkvist, Yeast-expressed Puumala hantavirus nucleocapsid protein induces protection in a bank vole model. Vaccine 20, 3523–3531 (2002)
J.D. Thompson, T.J. Gibson, F. Plewniak, F. Jeanmougin, D.G. Higgins, The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997)
T.A. Hall, BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acid Symp. Ser. 41, 95–98 (1999)
S. Braaker, G. Heckel, Transalpine colonisation and partial phylogeographic erosion by dispersal in the common vole Microtus arvalis. Mol. Ecol. 18, 2518–2531 (2009)
N. Saitou, M. Nei, The neighbor-joining method: a new method for reconstructing phylogentic trees. Mol. Biol. Evol. 4, 406–425 (1987)
S. Kumar, K. Tamura, M. Nei, Mega3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5, 150–163 (2004)
F. Ronquist, J.P. Huelsenbeck, MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572 (2003)
G.E. Schwarz, Estimating the dimension of a model. Ann. Stat. 6(2), 461–464 (1978)
D. Posada, iModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008)
K. Tamura, M. Nei, P.S. Kumar, Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl Acad. Sci. USA 101, 11030–11035 (2004)
M.A. Miller, M.T. Holder, R. Vos, P.E. Midford, T. Liebowitz, L. Chan, P. Hoover, T. Warnow, The CIPRES Portals. CIPRES, http://www.phylo.org/sub_sections/portal. Accessed 4 Aug 2009 (Archived by WebCite(r) at http://www.webcitation.org/5imQlJeQa)
J.C. Wilgenbusch, D.L. Warren, D.L. Swofford, AWTY: a system for graphical exploration of MCMC convergence in Bayesian phylogenetic inference (2004), http://ceb.csit.fsu.edu/awty
E.R. Kallio, J. Klingström, E. Gustafsson, T. Manni, A. Vaheri, H. Henttonen, O. Vapalahti, Å. Lundkvist, Prolonged survival of Puumala hantavirus outside the host: evidence for indirect transmission via the environment. J. Gen. Virol. 87, 2127–2134 (2006)
J. Hardestam, M. Simon, K.O. Hedlund, A. Vaheri, J. Klingström, Å. Lundkvist, Ex vivo stability of the rodent-borne Hantaan virus in comparison to that of arthropod-borne members of the Bunyaviridae family. Appl. Environ. Microbiol. 73, 2547–2551 (2007)
S. Schex, G. Dobler, J. Riehm, J. Müller, S. Essbauer, Rickettsia spp. in wild small mammals in Lower Bavaria, South-Eastern Germany. Vector Borne Zoonotic Dis. (2010) [Epub ahead of print]
R.G. Ulrich, J. Schmidt-Chanasit, M. Schlegel, J. Jacob, H.-J. Pelz, M. Mertens, M. Wenk, T. Büchner, D. Masur, K. Sevke, M.H. Groschup, F.-W. Gerstengarbe, M. Pfeffer, R. Oehme, W. Wegener, M. Bemmann, L. Ohlmeyer, R. Wolf, H. Zoller, J. Koch, S. Brockmann, G. Heckel, S.S. Essbauer, Network “Rodent-borne pathogens” in Germany: longitudinal studies on the geographical distribution and prevalence of hantavirus infections. Parasitol. Res. 103(Suppl. 1), 121–129 (2008)
M. Razzauti, A. Plyusnina, H. Henttonen, A. Plyusnin, Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 89, 1649–1660 (2008)
A. Dekonenko, V. Yakimenko, A. Ivanov, V. Morozov, P. Nikitin, S. Khasanova, T. Dzagurova, E. Tkachenko, C. Schmaljohn, Genetic similarity of Puumala viruses found in Finland and Western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. 3, 245–257 (2003)
J. Schmidt-Chanasit, S. Essbauer, R. Petraityte, K. Yoshimatsu, K. Tackmann, F.J. Conraths, K. Sasnauskas, J. Arikawa, A. Thomas, M. Pfeffer, J.J. Scharninghausen, W. Splettstoesser, M. Wenk, G. Heckel, R.G. Ulrich, Extensive host sharing of Central European Tula virus. J. Virol. 84, 459–474 (2010)
P. Kotlik, V. Deffontaine, S. Mascheretti, J. Zima, J.R. Michaux, J.B. Searle, A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc. Natl Acad. Sci. USA 103, 14860–14864 (2006)
G. Gerlach, K.F. Musolf, Fragmentation of landscape as a cause for genetic subdivision in bank voles. Conserv. Biol. 14(4), 1066–1074 (2000)
B. Walser, G. Heckel, Microsatellite markers for the common vole (Microtus arvalis) and their cross-species utility. Conserv. Genet. 9, 479–481 (2008)
M. Schweizer, L. Excoffier, G. Heckel, Fine-scale genetic structure and dispersal patterns in the common vole (Microtus arvalis). Mol. Ecol. 16, 2463–2473 (2007)
G. Heckel, R. Burri, S. Fink, J.-F. Desmet, L. Excoffier, Genetic structure and colonization processes in European populations of the common vole, Microtus arvalis. Evolution 59, 2231–2242 (2005)
V. Deffontaine, R. Libois, P. Kotlik, R. Sommer, C. Nieberding, E. Paradis, J.B. Searle, J.R. Michaux, Beyond the Mediterranean peninsulas: evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol. Ecol. 14, 1727–1739 (2005)
V. Deffontaine, R. Ledevin, M.C. Fontaine, J.P. Quéré, S. Renaud, R. Libois, J.R. Michaux, A relict bank vole lineage highlights the biogeographic history of the Pyrenean region in Europe. Mol. Ecol. 18, 2489–2502 (2009)
Å. Lundkvist, H. Meisel, D. Koletzki, H. Lankinen, F. Cifire, A. Geldmacher, C. Sibold, P. Gött, A. Vaheri, D.H. Krüger, R. Ulrich, Mapping of B-cell epitopes in the nucleocapsid protein of Puumala hantavirus. Viral Immunol. 15, 177–192 (2002)
K. Yoshimatsu, J. Arikawa, M. Tamura, R. Yoshida, Å. Lundkvist, B. Niklasson, H. Kariwa, I. Azuma, Characterization of the nucleocapsid protein of Hantaan virus strain 76–118 using monoclonal antibodies. J. Gen. Virol. 77, 695–704 (1996)
K.B. Sundström, M. Stoltz, N. Lagerqvist, Å. Lundkvist, K. Nemirov, J. Klingström, Characterization of two substrains of Puumala virus that show phenotypes that are different from each other and from the original strain. J. Virol. 85, 1747–1756 (2011)
D. Samuel, K. Sasnauskas, L. Jin, A. Gedvilaite, R. Slibinskas, S. Beard, A. Zvirbliene, S.A. Oliveira, J. Staniulis, B. Cohen, D. Brown, Development of a measles specific IgM ELISA for use with serum and oral fluid samples using recombinant measles nucleoprotein produced in Saccharomyces cerevisiae. J. Clin. Virol. 28, 121–129 (2003)
R. Slibinskas, D. Samuel, A. Gedvilaite, J. Staniulis, K. Sasnauskas, Synthesis of the measles virus nucleoprotein in yeast Pichia pastoris and Saccharomyces cerevisiae. J. Biotechnol. 107, 115–124 (2004)
M. Juozapaitis, A. Serva, A. Zvirbliene, R. Slibinskas, J. Staniulis, K. Sasnauskas, B.J. Shiell, L.F. Wang, W.P. Michalski, Generation of henipavirus nucleocapsid proteins in yeast Saccharomyces cerevisiae. Virus Res. 124, 95–102 (2007)
M. Juozapaitis, A. Serva, I. Kucinskaite, A. Zvirbliene, R. Slibinskas, J. Staniulis, K. Sasnauskas, B.J. Shiell, T.R. Bowden, W.P. Michalski, Generation of menangle virus nucleocapsid-like particles in yeast Saccharomyces cerevisiae. J. Biotechnol. 130, 441–447 (2007)
M. Juozapaitis, A. Zvirbliene, I. Kucinskaite, I. Sezaite, R. Slibinskas, M. Coiras, F. de Ory Manchon, M.R. López-Huertas, P. Pérez-Breña, J. Staniulis, I. Narkeviciute, K. Sasnauskas, Synthesis of recombinant human parainfluenza virus 1 and 3 nucleocapsid proteins in yeast Saccharomyces cerevisiae. Virus Res. 133, 178–186 (2008)
R. Petraityte, P.L. Tamosiunas, M. Juozapaitis, A. Zvirbliene, K. Sasnauskas, B. Shiell, G. Russell, J. Bingham, W.P. Michalski, Generation of Tioman virus nucleocapsid-like particles in yeast Saccharomyces cerevisiae. Virus Res. 145, 92–96 (2009)
P.J. Padula, C.M. Rossi, M.O. Della Valle, P.V. Martinez, S.B. Colavecchia, A. Edelstein, S.D.L. Miguel, R.D. Rabinovich, E.L. Segura, Development and evaluation of a solid-phase enzyme immunoassay based on Andes hantavirus recombinant nucleoprotein. J. Med. Microbiol. 49, 149–155 (2000)
R. Petraityte, L. Jin, R. Hunjan, A. Razanskiene, A. Zvirbliene, K. Sasnauskas, Use of Saccharomyces cerevisiae-expressed recombinant nucleocapsid protein to detect Hantaan virus-specific immunoglobulin G (IgG) and IgM in oral fluid. Clin. Vaccine Immunol. 14, 1603–1608 (2007)
R. Petraitytė, H. Yang, R. Hunjan, R. Ražanskienė, P. Dhanilall, R.G. Ulrich, K. Sasnauskas, L. Jin, Development and evaluation of serological assays for detection of Hantaan virus-specific antibodies in human sera using yeast-expressed nucleocapsid protein. J. Virol. Methods 148, 89–95 (2008)
K. Brus Sjölander, F. Elgh, H. Kallio-Kokko, O. Vapalahti, M. Hägglund, V. Palmcrantz, P. Juto, A. Vaheri, B. Niklasson, Å. Lundkvist, Evaluation of serological methods for diagnosis of Puumala hantavirus infection (nephropathia epidemica). J. Clin. Microbiol. 35, 3264–3268 (1997)
Acknowledgements
We would like to thank Ausra Razanskiene and Kestutis Sasnauskas (Vilnius) and Bernd Köllner (Riems) for continous support and helpful discussions, Åke Lundkvist (Stockholm), Kumiko Yoshimatsu and Jiro Arikawa (Sapporo) for provision of N protein-specific mAbs and Olli Vapalahti (Helsinki), Regina Allwinn and Holger F. Rabenau (Frankfurt) for provision of human serum panels. We kindly acknowledge Harald Weber, Aileene Lorber, Rahime Terzioglu, Elmar Schröpfer, Ralf Hagen, Mirko Köhler, Anne Grumbach, Peter Klein and Kerstin Weiss for their support during rodent trapping, rodent necropsy and collection of human sera in South-East Germany. The excellent technical lab assistance of Jana Blumhardt, Ina Römer, Dörte Kaufmann, Sabine Fink and Astrid Thomas is gratefully acknowledged. The work was supported by Research-Project M/SAB1/5/A017 for the Bundeswehr Medical Service to RGU.
Author information
Authors and Affiliations
Corresponding author
Additional information
Marc Mertens and Eveline Kindler contributed equally to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11262_2011_620_MOESM1_ESM.pptx
Supplementary Figure. Bayesian reconstruction of phylogenetic relationships based on 968 bp mtDNA (cyt b) from 120 Myodes glareolus with M. rufocanus as outgroup. Only support values for main nodes that connect major evolutionary lineages are displayed. Posterior probabilities are indicated above the major branches and percentage of bootstrap support for neighbour-joining (NJ) algorithms below the branches. * indicates a different topology based on NJ algorithms, ns refers to posterior probabilities <0.50 and percentages of bootstrap support <50%. Arrows depict cyt b sequences from bank voles from Bavaria where PUUV sequences were detected in (PPTX 64 kb)
Rights and permissions
About this article
Cite this article
Mertens, M., Kindler, E., Emmerich, P. et al. Phylogenetic analysis of Puumala virus subtype Bavaria, characterization and diagnostic use of its recombinant nucleocapsid protein. Virus Genes 43, 177–191 (2011). https://doi.org/10.1007/s11262-011-0620-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-011-0620-x