
Abstract
The broad distribution and prevalence of H3 subtype influenza viruses in avian and mammalian hosts constitutes a global threat to both human and veterinary health. In this present study, six H3N8 influenza viruses isolated from domestic ducks during 2004–2005 in northern China were genetically and phylogenetically characterized. Sequence analysis showed that HA, NA, and M genes of all the six H3N8 isolates had a close relationship with those of Equine/Jilin/1/89 (H3N8) virus, which once caused outbreak in equine populations in northern China. The PB2 and PA genes of the viruses possessed the highest similarities with highly pathogenic avian H5N1 influenza viruses currently circulating in this region. These findings emphasize the importance of avian influenza virus surveillance in this region for understanding the genesis and emergency of novel reassortants with pandemic potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wild aquatic birds are regarded as the natural reservoirs of influenza A viruses [1], and domestic ducks play an important role in the transmission of influenza virus from wild aquatic birds to terrestrial poultry [2, 3]. Currently, 16 HA and 9 NA have been identified from aquatic birds [4, 5]. However, one of the most frequently isolated subtypes is H3 subtype influenza viruses [6, 7], with its wide host range including pigs, horses, and humans [8]. Historically, H3N8 caused more severe diseases to horses and A/Duck/Ukraine/1/63 (H3N8) was presumed to donate HA and PB1 genes to the 1968 Hong Kong H3N2 pandemic strain [9, 10]. In addition to the classic form of H3N8 found to infect horses since 1977, another independent interspecies transmission of non-reassortant H3N8 virus directly from avian to equines caused two consecutive outbreaks in northeast China in 1989 and the following year [11]. Recent H3N8 outbreaks in horses in Japan, Australia, and China resulted in large economic losses [12].
In this study, six H3N8 influenza viruses isolated from domestic ducks during 2004–2005 in northern China were genetically and phylogenetically characterized.
Materials and methods
Viruses
Six H3N8 influenza viruses were isolated from cloacal swabs of clinically healthy ducks in different live bird markets in Beijing during 2004–2005. Initial isolations of the viruses were performed in 10-day-old specific pathogen free (SPF) embryonated chicken eggs (ECE) at 35°C for 48 h. Subtype identification of the viruses were determined by standard hemagglutination inhibition and neuraminidase inhibition assays using specific antisera to the reference strains of influenza viruses [13]. Allantoic fluid were harvested from ECE second passage viruses and stored at −70°C. The six viruses in this study were designated as: A/Duck/Beijing/33/04 (Dk/BJ/33/04), A/Duck/Beijing/40/04 (Dk/BJ/40/04), A/Duck/Beijing/44/04 (Dk/BJ/44/04), A/Duck/Beijing/56/05 (Dk/BJ/56/05), A/Duck/Beijing/59/05 (Dk/BJ/59/05), A/Duck/Beijing/61/05 (Dk/BJ/61/05).
RNA extraction and RT-PCR
Viral RNA was extracted from allantoic fluid by using TRIzol reagents (Gibco-BRL) and reverse transcription was performed by using oligonucleotide influenza universal primer Uni12: 5′-AGC AAA AGC AGG-3′. After reverse transcription, PCR was done as described previously [14], using primers (sequences available on request) specific for each of the eight RNA segments.
Gene sequence
PCR products were purified with a QIAquick PCR purification kit (Qiagen). The purified PCR products were then partially sequenced by using an Amersham ET Dye terminator kit and analyzed with an ABI 3730 DNA sequencer (Perkin–Elmer Applied Biosystems). Assembly of sequences, translation of nucleotide sequences into protein sequences, and initial multiple sequence alignments were performed with the CLUSTAL_V method using MegAlign software version 1.03 (DNAStar Inc.).
Phylogenetic analysis
Phylogenetic analysis was carried out by analyzing the data obtained here with those of other sequences of influenza viruses from GenBank database. A neighbor-joining nucleic acid tree was constructed in MEGA 3.1 using the Kimura 2-parameter model with 1,000 bootstrap replicates as previously described [15, 16]. In this study, the nucleotide sequences used for the phylogenetic analysis are as follows: PB2 (nt 1345–2202), PB1 (nt 1181–1814), PA (nt 630–1229), HA (nt 65–1054), NP (nt 637–1263), NA (nt 35–1420), M (nt 111–731), NS (nt 60–716).
Nucleotide sequence accession numbers
The nucleotide sequences for all H3N8 influenza viruses determined in this present study are available from GenBank under accession numbers EU492487 to EU492534.
Results
Genetic analysis
Hemagglutinin (HA) is one of the surface glycoproteins, which mediates viral entry into host cells and elicits the production of neutralizing antibody. In this study, full length HA gene sequences were determined. The HA genes of the six isolates showed 99.93% nucleotide sequence homology. HA genes of the recent H3N8 viruses, A/equine/Kanazawa/1/2007 and A/equine/Mongolia/1/2008, which caused horse disease in Japan and Mongolia, had 77.15% and 74.52% similarity with those of these isolates, respectively. However, HA genes of these isolates possessed 94.86% similarity with that of Equine/Jilin/1/89 (H3N8) virus, which caused two consecutive outbreaks in northeast China in 1989 and 1990. The results of this study indicated that the HA genes of the early equine virus were still maintained in avian hosts.
Five of the H3N8 viruses shared the same amino acid sequence of -PEKQTR/G- at the cleavage site between HA1 and HA2, which was same as those of other reported avian H3 viruses. DK/BJ/61/05 possessed -PEKQPR/GL- at the cleavage site (Table 1). The cleavage site sequences of these viruses were characteristic of LPAI viruses [17]. Analysis of the potential glycosylation sites (PGS) of the H3N8 isolates revealed that they were indistinguishable, with five PGS in HA1 (8,22,38,165,285) and one in HA2 (154).
Avian type amino acid residues can be observed in these viruses at the receptor binding sites of HA gene at positions 183 (H), 190 (E), and 226 (Q) (Table 1). Compared with Dk/Ukraine/1/63 (H3N8), the proposed donor of 1968 Hong Kong pandemic strain, these viruses had two amino acid residues’ difference at positions 137 and 227. In addition, the mammalian-adapted mutation PB2 E627K was not found in the six isolates.
Phylogenetic analysis
To determine the molecular evolution of the six domestic duck H3N8 viruses, phylogenic analysis of the eight segments were carried out. In the analysis, H3 and other subtype avian influenza viruses from different regions and periods were selected.
Surface genes (HA and NA)
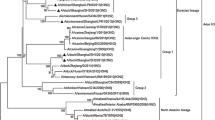
Phylogenically, the HA genes of H3 subtype viruses were phylogenically divided into American and Eurasian lineages (Fig. 1a). The Eurasian lineage formed two groups: I and II. The six isolates mapped to group II along with Dk/Ukr/1/63(H3N8), Equine/Jilin/1/89(H3N8) and most H3 avian influenza viruses from China and Korea. The fact that Dk/Ukr/1/63(H3N8) and other earlier avian H3 virus strains clustered with recent avian H3 isolates indicates that the evolutionary rate of H3 HA gene in avian species is relatively slow, and the Ukraine-like H3 genes is still the prevalent genotype in China. While the group II viruses were mainly composed of Chinese viruses, the viruses in the group I were from more widely geographic regions, such as Russia, Japan, Italy, and China. It is presumed that different transmission modes exist among these two groups: long-distance flights of migratory birds may play an important role in the transmission of group I, and local poultry trade accounted for the virus spread in group II.


Phylogenetic trees for the HA (a), NA (b), PB2 (c), PB1 (d), PA (e), NP (f), M (g), and NS (h) genes of the H3N8 avian influenza viruses. The HA, PB1 and PA phylogenetic trees are rooted to A/Equine/London/1416/73 (H7N7) and those of PB2, NP, NA, M, and NS to A/Equine/Praque/1/56 (H7N7). Viruses in the present study are in italic and bold. Ck chicken, Dk duck, Gs goose, HK Hong Kong
Like HA, the NA phylogenic tree was also divided into American and Eurasian lineages (Fig. 1b). The six H3N8 viruses formed an independent branch. The Eurasian lineage was further divided into two groups, A and B. Six H3N8 viruses, Dk/Ukr/1/63, Equine/Jilin/1/89 were located in group B along with viruses from China and the neighboring regions of Hong Kong, Korea, Japan, and Australia. But the viruses in the group A were mostly from distant regions, such as the Netherlands, Norway, and South Africa. These findings also supported the presumption that migratory birds and poultry trade might play important roles in transmission patterns of avian H3 influenza viruses in nature.
Internal genes
Phylogenic analysis of internal genes showed that all six isolates belonged to the Eurasian lineage (Fig. 1c–g), and the similarity of these internal genes ranged from 97.16% to 100%. NS genes were divided into alleles A or B, and the present viruses were located in allele B (Fig. 1h).
The internal genes of all the six isolates formed independent branches within the phylogenic trees except for the M and NS genes where in both instances the viruses from 2004 formed distinct branches from those of 2005 (Fig. 1g, h). In the M gene tree, the viruses isolated during 2004 and 2005 had the highest nucleotide sequence homology with Duck/Jiangxi/2374/05 (H3N6) and Equine/Jilin/1/89 (H3N8), respectively (Fig. 1g). In the NS gene tree, the 2004 isolates had high homology with Goose/Guiyang/3799/05(H5N2), and the 2005 isolates had close relationship with Dk/Yangzhou/02/05 (H8N4) (Fig. 1h).
It was noteworthy that extensive reassortment in the internal genes between the H3N8 isolates and other subtype influenza viruses can be observed. In particular, the PB2 and PA genes of the six isolates possessed the highest similarity with those of recently isolated H5N1 viruses (Fig. 1c, e). The PB1 genes of the 2004 and 2005 H3N8 viruses also formed an independent branch from other H3 viruses (Fig. 1d). Interestingly, the NP genes of the six isolates had the highest identities with the earlier European HPAI viruses Chicken/Italy/312/97 (H5N2), Turkey/England/50-92/91 (H5N1) and LPAI virus Duck/Germany/113/95 (H9N2) (Fig. 1f).
In summary, the phylogenic analysis of the 2004 and 2005 H3N8 viruses indicated that the surface genes possessed a close relationship with that of early Dk/Ukraine/1/63-like or Equine/Jilin/1/89-like viruses. Results of internal genes showed reassortment events occurred, probably in domestic ducks, between these H3 subtype viruses and other subtype viruses.
Discussion
Influenza viruses possessing the H3 hemagglutinin are one of the most frequently isolated subtypes from feral ducks and also the major subtype that caused human disease. Previous study has shown that a virus similar to the duck virus, Dk/Ukraine/1/63 (H3N8), donated the HA and PB1 genes to the 1968 Hong Kong pandemic strain. Since the outbreaks of HPAI H5N1 in poultry in Southern China in 1996 [18] and the occurrence of human H5N1 infection incident in 1997 [19], extensive surveillance shows aquatic birds, especially domestic ducks, are even becoming the perpetuation host of H5N1 viruses [2]. The fact that the H5N1 viruses that caused poultry disasters and occasional human infection, and H3 virus that historically caused pandemic were found together in duck populations raised the possibility that these viruses could reassort to change the biology of H3 influenza virus in domestic ducks. The aim of this present study was to genetically and phylogenetically characterize the H3N8 isolates from domestic duck.
Phylogenetic analysis indicated that the HA and NA genes of the six H3N8 duck influenza viruses continued to be Dk/Ukraine/1/63-like, with the evolution of the two surface glycoproteins’ genes being conservative in ducks. Meanwhile, the six isolates had close relationship with Equine/Jilin/1/89 (H3N8), which caused severe disease in horses with 20% mortality in Northern China during 1989–1990 and disappeared in the following year. The close phylogenic relationship of these viruses revealed that these H3N8 viruses might still have the possibility to infect horses, and the biosecurity measures should be implemented for horse population.
The phylogenic analysis of internal genes showed that the PB2 and PA genes of these isolates had the highest homology with recent H5N1 viruses from China, indicating these two gene segments might be derived from H5N1 viruses by reassortment. This was consistent with several recent reports of reassortments in the genesis of recent H3 avian influenza variants [20, 21]. The H3 virus causing outbreaks in chicken flocks in Italy was also a reassortant [22]. The latest report showed that a new subtype virus Ck/Kor/164/04 (H9N8) whose eight segments came from aquatic bird H3N8 virus and chicken H9 virus, respectively, caused 20% mortality in the infected chickens [23]. All these facts remind us that the genetic reassortment among avian influenza viruses can facilitate the virus variation and the emergence of novel strains. Therefore, LPAI viruses like H3 subtype viruses as well as HPAI viruses should be monitored closely. In addition, the internal genes of the six H3N8 isolates from 2004 and 2005 showed different evolutionary trends from other H3 viruses in China. These six viruses clustered together and formed an independent branch distinct from other H3 viruses especially with respect to the internal genes, suggesting that multiple genotypes H3 influenza viruses are circulating in China [20].
The fact that the HA and NA genes of the H3N8 viruses prevailing in China maintained to be Dk/Ukraine/1/63-like or Equine/Jilin/1/89-like indicated the continuing threat to both the veterinary and human health. In particular, the transmissibility and pathogenicity of these H3N8 viruses resulted from reassortment with HPAI H5N1 deserve more in-depth investigation.
References
R.G. Webster, W.J. Bean, O.T. Gorman, T.M. Chambers, Y. Kawaoka, Microbiol. Rev. 56, 152–179 (1992)
D.J. Hulse-post, K.M. Sturm-ramirez, J. Humerd, P. Seiler, E.A. Govorkova, S. Krauss, C. Scholtissek, P. Puthavathana, C. Buranathai, T.D. Nguyen, H.T. Long, T.S. Naipospos, H. Chen, T.M. Ellis, Y. Guan, J.S. Peiris, R.G. Webster, Proc. Natl Acad. Sci. USA 102, 10682–10687 (2005). doi:https://doi.org/10.1073/pnas.0504662102
Y. Kawaoka, Influenza Virology: Current Topics, 1st edn. (Arnold. Caister Academic, London, 2006)
A.T. Baker, J.N. Varghese, W.G. Laver, G.M. Air, P.M. Colman, Proteins 2, 111–117 (1987). doi:https://doi.org/10.1002/prot.340020205
R.A.M. Fouchier, V. Munster, A. Wallensten, T.M. Bestebroer, S. Herfst, D. Smith, G.F. Rimmelzwaan, B. Olsen, A.D. Osterhaus, J. Virol. 79, 2814–2822 (2004). doi:https://doi.org/10.1128/JVI.79.5.2814-2822.2005
J. Pasick, H. Weingartl, A. Clavijo, J. Riva, H. Kehler, K. Handel, E. Watkins, K. Hills, Avian Dis. 47, 1208–1213 (2003)
E.F. Kaleta, G. Hergarten, A. Yilmaz, Dtsch. Tierarztl. Wochenschr. 112(12), 448–456 (2005)
W.J. Bean, M. Schell, J. Katz, Y. Kawaoka, C. Naeve, O. Gorman, R.G. Webster, J. Virol. 66, 1129–1138 (1992)
C.W. Ward, T.A. Dopheide, Biochem. J. 195, 337–340 (1981)
R. Fang, W. Min Jou, D. Huylebroeck, R. Devos, W. Fiers, Cell 25, 315–323 (1981). doi:https://doi.org/10.1016/0092-8674(81)90049-0
Y. Guo, M. Wang, Y. Kawaoka, O. Gorman, T. Ito, T. Saito, R.G. Webster, Virology 188(1), 245–255 (1992). doi:https://doi.org/10.1016/0042-6822(92)90754-D
Anonymous, World Animal Health Information Database (WAHID)—Weekly Disease Information (http://www.oie.int/wahid-prod/public.php?page=weekly_report_index&admin=0)
A.P. Kendal, M.S. Pereira, J.J. Skehel, Concepts and Procedures for Laboratory-Based Influenza Surveillance (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, 1982)
L.L. Shu, Y.P. Lin, S.M. Wright, K.F. Shortridge, R.G. Webster, Virology 202, 825–833 (1994). doi:https://doi.org/10.1006/viro.1994.1404
S. Kumar, K. Tamura, M. Nei, Brief. Bioinform. 5, 150–163 (2004). doi:https://doi.org/10.1093/bib/5.2.150
M.D. Saad, L.S. Ahmed, M.A. Gamal-Eldein, M.K. Fouda, F.M. Khalil, S.L. Yingst, M.A. Parker, M.R. Montevillel, Emerg. Infect. Dis. 13(7), 1120–1121 (2007)
D.A. Steinhauer, Virology 258, 1–20 (1999). doi:https://doi.org/10.1006/viro.1999.9716
X. Xu, K. Subbarao, N. Cox, Virology 261, 15 (1999). doi:https://doi.org/10.1006/viro.1999.9820
K. Subbarao, A. Klimov, J. Katz, H. Regnery, W. Lim, H. Hall, M. Perdue, D. Swayne, C. Bender, J. Huang, M. Hemphill, T. Rowe, M. Shaw, X. Xu, K. Fukuda, N. Cox, Science 279, 393–396 (1998). doi:https://doi.org/10.1126/science.279.5349.393
M. Liu, S.Q. He, W. David, N.N. Zhou, D.R. Perez, M. Bing, L. Fan, X.T. Huang, R.G. Webster, R.J. Webby, Virology 305, 267–275 (2003). doi:https://doi.org/10.1006/viro.2002.1762
Y.K. Choi, S.H. Seo, J.A. Kim, R.J. Webby, R.G. Webster, Virology 332, 529–537 (2005). doi:https://doi.org/10.1016/j.virol.2004.12.002
L. Campitelli, C. Fabiani, S. Puzelli, A. Fioretti, E. Foni, A.D. Marco, S. Krauss, R.G. Webster, I. Donatelli, J. Gen. Virol. 83, 413–420 (2002)
Y.J. Lee, J.Y. Shin, M.S. Song, Y.M. Lee, J.G. Choi, E.K. Lee, O.M. Jeong, H.W. Sung, J.H. Kim, Y.K. Kwon, J.H. Kwon, C.J. Kim, R.J. Webby, R.G. Webster, Y.K. Choi, Virology 359, 313–323 (2007). doi:https://doi.org/10.1016/j.virol.2006.09.025
Acknowledgments
This study was supported by the National Natural Scientific Foundation (30599431), National Key Technologies R&D Program (2006BAD06A01), National Basic Research Program (973) (2005CB523003), 863 program (2006AA10A205), EU AIV project FLUINNATE (SP5B-CT-2006-044161) and CAU Scientific Research Initiation Foundation (2008016). J. H. Liu was funded by Taishan Scholar Foundation. E. G. Brown was funded by CIHR Grant number TPA90188.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pu, J., Liu, QF., Xia, YJ. et al. Genetic analysis of H3 subtype influenza viruses isolated from domestic ducks in northern China during 2004–2005. Virus Genes 38, 136–142 (2009). https://doi.org/10.1007/s11262-008-0300-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0300-7