
Abstract
The complete genome of the Chilean isolate Cl-766 of Grapevine leafroll-associated virus-3 (GLRaV-3) has been sequenced. This is the first genome sequence obtained from a GLRaV-3 isolate of the Southern hemisphere. The genomic RNA of 17,919 nucleotides contains 13 open reading frames (ORFs) with 5′ and 3′ untranslated regions (UTR) of 158 and 277 nucleotides, respectively. Comparison with NY1, the only isolate with complete genomic sequence available today, shows 97.6% nucleotide identity between the two isolates. Examination of the genome variability shows that most of the genetic diversity is concentrated in ORF1a. Three additional isolates from different geographic regions of Chile were partially sequenced as well, one which showed sequence divergence with respect to the other local and foreign isolates, indicative of different evolutionary constrains. Immunodetection systems were developed using monoclonal and polyclonal antibodies produced against the recombinant major coat protein of GLRaV-3, providing sensitive and specific detection using a triple antibody sandwich–enzyme linked immunosorbent assay (TAS-ELISA) and an immunocapture-reverse transcription-polymerase chain reaction (IC-RT-PCR) assay.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grapevine leafroll-associated virus-3 (GLRaV-3) causes important economical losses in Chile and the world [1, 2]. This virus belongs to the family Closteroviridae and is classified under the genus Ampelovirus [3]. Several serologically distinct viruses within this family have been associated with leafroll disease, and all of them are characterized by the presence of long filamentous viral particles [4]. These viruses exhibit the most complex regulation among the alphavirus-like superfamily, using different expression strategies, such as subgenomic RNAs, polyprotein processing, and predicted translational frameshifting [5].
The GLRaV-3 genome consists of a linear monopartite, positive-sense single-stranded RNA. GLRaV-3 is phloem limited, infects only dicotyledonous hosts and is transmitted semi-persistently by coccid or pseudococcid mealybug vectors [3, 6]. Until now, only one isolate (NY1, from USA) has been fully sequenced and limited information is available for other isolates for which only partial sequences have been reported [7]. Moreover, only three additional members of the Ampelovirus genus have recently been completely sequenced, the Little cherry virus-2 (LChV-2) isolate USA6b of 15 kb [8], one isolate of Plum bark necrosis stem pitting-associated virus (PBNSPaV) of 14.2 kb [9] and one isolate of Pineapple mealybug wilt-associated virus-1 (PMWaV-1) of 13.1 kb (GenBank accession AF 414119). GLRaV-3 contains the second largest genome among sequenced closteroviruses after Citrus tristeza virus (CTV) of 19.3 kb which has the largest genome of all known positive strand plant RNA viruses [7, 10].
Cabaleiro and Segura reported an increase from 33% to 83% in the GLRaV-3 incidence during 5 years in a vineyard of northwestern Spain [6]. Another study reported an increase from 11% to 90% in New Zealand vineyards during the same period of time [11]. As it might be expected from these reports, spreading of the virus must be controlled by propagating certified material and controlling vectors in the field. Both strategies require efficient viral detection systems.
In the present article, we report the complete genome sequence of a Chilean GLRaV-3 isolate. Sequence analyses and phylogenetic studies of the GLRaV-3 genome, compared with other local and foreign isolates are also reported. This contribution represents the second complete GLRaV-3 genome and the first sequenced from the Southern hemisphere. We also describe specific and sensitive methods for detection of the virus including triple antibody sandwich–enzyme linked immunosorbent assay (TAS-ELISA) and immunocapture-reverse transcription-polymerase chain reaction (IC-RT-PCR) using antibodies raised against the recombinant major coat protein.
Materials and methods
Viral source and total RNA isolation
Naturally infected Merlot grapevine isolates Cl-664, Cl-765, and Cl-766 from the VI region, and Chardonnay grapevine isolate Cl-817 from the Metropolitana Region of Chile were used. Total RNA was extracted from fresh bark scrapings grinded in liquid nitrogen according to Chang et al. [12].
Reverse transcription-polymerase chain reaction (RT-PCR) and cloning
Approximately, 500 ng of grapevine total RNA was heated for 10 min in the presence of 50 ng of random hexamers (Invitrogen) and reverse transcribed in a total volume of 25 μl for 1 h at 37°C with M-MLV reverse transcriptase (Promega). Five μl of the product were used in the PCR reaction with specific viral primers (Table 1) and Taq or Tli DNA polymerase (Promega). The PCR involved a 2-min heating step at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 55–59°C (depending on the primers pair), 1 min at 72°C, and a final step of 10 min at 72°C. For sequencing purposes, PCR products were purified with QIAquick PCR purification kit (Qiagen), cloned in pGEM-T vector (Promega) and used to transform E. coli Nova blue cells (Novagen). Sequencing was performed with an Applied Biosystems ABI 310 automated sequencer using M13 primers for all pGEM-T clones and additional specific primers (Table 1). In order to increase the reliability, at least two overlapping clones from different PCR products were sequenced each time.
Enzyme Linked Immunosorbent Assay (ELISA)
Grapevine samples were obtained by grinding 500 mg of fresh bark scrapings in 2 ml of extraction buffer according to a commercially available protocol (Bioreba). Sap extract was used as an antigen in DAS-ELISA (Double Antibody Sandwich–Enzyme Linked Immuno Sorbent Assay) or TAS-ELISA (Triple Antibody Sandwich–Enzyme Linked Immuno Sorbent Assay) [13, 14]. MaxiSorp ELISA plates (Nunc) were coated with a rabbit polyclonal antibody for 3 h at 37°C. After a washing step, plates were incubated overnight at 4°C with grapevine sap (infected or uninfected) or purified recombinant major coat protein. Plates were then washed and anti-coat protein monoclonal antibody was added to each well and incubated for 4 h at 37°C. After washing, 200 μl of rabbit anti-mouse IgG (H+L) alkaline phosphatase conjugated (Pierce) diluted 1:28,000 in conjugate buffer was added to each well and incubated at 37°C for 1 h (for TAS-ELISA only). Finally, 200 μl of the substrate p-nitrophenyl phosphate at 1 mg/ml was added to each well and incubated at 37°C for 1–2 h. Color development was monitored by measuring the absorbance at 405 nm after 30, 60, and 90 min. Antibodies from commercial kits (Bioreba) were included for validation of the tests. A reaction was considered positive when the mean absorbance for a well was at least three times the mean value of a sample from a non-infected control plant. All tests were repeated three times in duplicate wells.
Immunocapture-RT-PCR (IC-RT-PCR)
The IC-RT-PCR amplification protocol was derived from the procedure of Sefc et al. [15]. Microcentrifuge tubes were coated with an antibody mix containing rabbit polyclonal antibody (1:2,000) and the monoclonal antibodies 5A5/C2 and 8G5/H6 (1:4,000) diluted in NaHCO3 0.1 M pH 9.6 for 2 h at 37°C. After washing three times with PBS-0.05% Tween 20, coated tubes were incubated overnight at 4°C with 50 μl of plant sap, and then washed again. Later, the RT reaction mix was added directly to each tube and incubated for 1 h at 37°C in a total volume of 45 μl with M-MLV. For the PCR step, 10 μl of cDNA were used with specific viral primers LC1F and LC2R (Table 1) and Taq DNA polymerase. The PCR involved a heating step of 2 min at 94°C, followed by 38 cycles of 30 s at 94°C, 30 s at 58°C, 50 s at 72°C, and a final step of 10 min at 72°C. PCR products were sequenced to confirm virus detection.
GLRaV-3 genome analysis
Sequence analysis
Each GLRaV-3 ORF was annotated using Artemis [16]. GLRaV-3 ORFs with unknown function were analyzed with InterProScan [17] and compared with updated public available databases to find patterns and infer putative functions. The intergenic regions I1 (228 nucleotides) and I2 (1,065 nucleotides) located at genome coordinates 8,479–8,707 and 8,864–9,929, respectively were also analyzed. Databases and comparisons were performed using BLAST programs [18]. The length of the 5′ UTR and 3′ UTR extremities might be viewed with caution, since the viral ends were amplified with specific primers (see Table 1) without further analysis by RACE.
Genomic variability
The Chilean (EU344893) and the USA (NC004667) GLRaV-3 genomes were aligned and the variability for each position in the alignment was calculated with the Shannon formula [19] and graphically represented using BioEdit v7.0 [20]. The positions with higher entropy value were assumed as less conserved positions. A sequence coverage map was built by recursively adding all the available sequences using the alignment of both GLRaV-3 complete genomes as template. Chilean isolates Cl-766 (EU344893), Cl-765 (EU344896), Cl-664 (EU344895), and Cl-817 (EU344894) were also considered and sequence alignments were performed using ClustalX v1.83 [21].
Phylogenetic analysis
A phylogenetic study was performed using the alignment of the major coat protein sequences from several virus isolates. Instead of 12 complete coat proteins (CP) ORFs, a partial CP ORF alignment was done considering 28 sequences of 484 nucleotides obtained from the 5′ end (Fig. 2). Construction of the evolutionary model was performed using the Bayesian information criterion (BIC) included in the Modelgenerator software [22, 23] to guarantee that the complete CP and the partial CP ORFs share the same evolutionary model. Identical sequences were excluded from the analysis and the divergent CP (CPd) ORF sequence from isolate Cl-766 was included as outgroup. The phylogenetic tree was obtained with MEGA4 [24] using the Neighbor-joining method and 1,000 bootstrap iterations as a confidence test. The evolutionary distances were calculated using the Maximum Composite Likelihood method [24] and the codon positions included were 1st + 2nd + 3rd.
Viral protein expression in E. coli
The GLRaV-3 CP complete ORF from isolate Cl-766 was obtained by RT-PCR using primers LR3CPF1 and LR3CPR1 with BamHI and NotI restriction sites, respectively (Table 1) and then cloned in the pGEM-T vector. The CP was digested from pGEM-T and sub cloned in the vectors pET-32a (Novagen) and pGEX-6P-1 (Amersham). Recombinant clones were used to transform E. coli BL21(DE3) competent cells and the expression was induced by incubation with 1 mM IPTG during 3 h after 0.5 OD600 nm was reached. Insoluble fractions were solubilized and purified in the presence of 5 M urea for CP-TRX and 1% N-lauroylsarcosine for GST-CP. The histidine-tagged fusion protein was purified with a Ni-agarose column (Qiagen) [25] and the GST-tagged protein was purified by a glutathione-Sepharose column (Amersham) chromatography [26]. Protein expression was analyzed by SDS-PAGE [27].
Monoclonal antibody production
Two month old female Balb/c mice were immunized intraperitoneally three times at 3 week intervals with 50 μg of the purified viral recombinant major coat protein diluted in PBS and emulsified 1:1 with Freund complete adjuvant for the first injection, and incomplete Freund adjuvant for the second and third injections. Ten days after the last injection, animals were bled from the tail to obtain serum. Humoral response was determined by ELISA [13] and Western blot [28]. In order to produce the hybridoma, spleen cells from the immunized mice with the highest titer against the recombinant proteins were isolated and fused with NS0/2 mouse myeloma cells [29].
Polyclonal antibody production
Four months old female New Zealand white rabbits were immunized subcutaneously five times at 15-day intervals. Each injection contained 500 μg of the purified viral recombinant major coat protein diluted in PBS and emulsified 1:1 with Freund complete adjuvant for the first injection, and incomplete Freund adjuvant for the remaining injections. After the third injection, rabbits were bled weekly to evaluate serum titers by ELISA and Western blot.
Results and discussion
Sequence analysis and genome organization of GLRaV-3
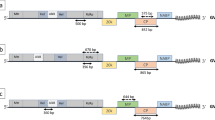
Despite that GLRaV-3 is widely spread in Chile [30], no sequence information was available from a Chilean isolate. Until now, only one full-length GLRaV-3 genomic sequence (NY1 from USA) has been reported [7]. In the present work, the genome of GLRaV-3 isolate Cl-766 (EU344893) was cloned and entirely sequenced. For this, primers mainly based on the NY1 (NC004667) sequence were used to obtain overlapping RT-PCR products with sizes ranging from 546 bp to 1,133 bp (Table 1). Comparisons to the NY1 isolate genome showed a 97.6% of nucleotide sequence identity. Additionally, the genome of isolates Cl-664, Cl-765, and Cl-817 were partially sequenced to cover 8% of its size (Fig. 1). This included fragments from clones A and H containing part of HSP70h ORF, and the complete CP coding region (Fig. 1).

Grapevine leafroll-associated virus-3 genome organization and sequencing strategy. Schematic representation of the selected overlapping segments used to determine the sequence of the genome of Chilean GLRaV-3 isolates Cl-766, Cl-817, Cl-765, and Cl-664
The genome organization of isolate Cl-766 resembles that of NY1 isolate, with a length of 17,919 nucleotides encompassing 13 ORFs and 5′ and 3′ UTRs of 158 and 277 nucleotides, respectively.
The first AUG codon of the viral polyprotein ORF1a starts at nucleotide 159. This ORF encodes for a papain-like proteinase (Pro), a methyltransferase (MET), an AlkB domain (detailed below), and a helicase (HEL). ORF1b encodes for a RNA-dependant RNA polymerase (RdRp) that is presumable expressed by means of a predicted +1 translational frameshifting between ORF1a and ORF1b [31]. Previous studies have demonstrated in Beet yellow closterovirus (BYV) that the ectopic expression of MET-HEL generates both separated proteins, suggesting that the MET-HEL polyprotein could be processed by a still unknown protease [32, 33]. Supporting these results, there is a strong evidence to suggest that CTV RdRp is cleaved by a host protease [34]. ORFs 1a and 1b together with 5′ and 3′UTRs are sufficient to replicate CTV genome in Nicotiana benthamiana protoplasts [35].
The above assembled segments, together with all available GLRaV-3 sequences, were mapped onto the genome coordinates to illustrate the current sequence distribution and availability in GenBank. As it can be seen in Fig. 2, a heterogeneous distribution of sequenced segments covers the GRLaV-3 genome. Examination of Fig. 2 shows that the parts of RdRp, HSP70h, and CP ORFs appear highly represented by 16, 27, and 33 sequences, respectively. The coordinates with lowest representation were located in part of the polyprotein and the intergenic fragment I2, with only two non-redundant sequences available. Notably, more than 65% of the whole genome is solely covered by 2–3 sequences (depending on the region) including those reported in the present article (Fig. 2).

Sequence coverage of the genomic fragments available for GLRaV-3 at public databases including Chilean isolates Cl-766, Cl-817, Cl-765, and Cl-664 sequenced in this work
An entropy plot reflecting the variability for each position in the alignment of the two complete GLRaV-3 genomes available is presented in Fig. 3. We used the complete genome sequences instead of all the available partial fragments to avoid bias due to the different number of sequences available for each genome region. Dark bars denote higher variability, which is mainly concentrated in the polyprotein ORF1a between nucleotides 1,070–3,700 and 4,330–4,740. These results agree with others previously described by Ling et al. [7], who reported more changes in the first 4,765 nucleotides than in the rest of the genome. Different results were obtained by Little et al. [36] with GLRaV-1, another member of the genus Ampelovirus. Using a 100 bp window, they observed a different variability distribution in 10 ORFs (12.4 Kb) of GLRaV-1 genome, with the highest variations located in HSP70h, CPd1, and CPd2 ORFs.

Entropy plot covering the whole genome alignment between NY1 (NC004667) and Cl-766 (EU344893) GLRaV-3 isolates. The verticals bars indicate coordinates where nucleotides changes occur
Considering that the MET-HEL-RdRp module of the closteroviruses genomes is universally conserved within the alphavirus-like superfamily, less variability was expected especially for the variable MET coding segment. In contrast, the large space of more than 3,000 nucleotides between the MET and HEL coding segments corresponding to more than 15% of the whole genome and with a function still unclear is highly variable. The variable segments located through the MET region extend to the upstream nucleotides of the recently described AlkB domain (Fig. 3). AlkB-like proteins are members of the 2-oxoglutarate and Fe(II)-dependent oxygenase superfamily. Importantly, the AlkB domain was found between aa 1,618 and 1,691 in ORF1 of isolate Cl-766, as well as the specific motifs required for its function. This domain, which could be involved in protecting the virus against post-transcriptional gene silencing (PTGS) or in viral RNA repair caused by environmental methylating compounds via methylation reversal, has been reported in 22 RNA positive-strand plant viruses, but only in two ampeloviruses, GLRaV-3, and LChV-2 [32, 37].
Interestingly, members of the genera Closterovirus and Mimivirus are only reported to contain a HSP70h gene [32]. A central question that remains unsolved is the complexity shared by these two viruses considering that they belong to different taxa and infect different hosts. Moreover, mimiviruses exhibit a huge 1.2 megabase double-stranded DNA genome [38] compared to the 18 kb of the GLRaV-3 genome.
The genome of isolate Cl-766, similar to the genome of isolate NY1, contains the same two non-coding intergenic regions I1 and I2 of 228 and 1,065 nucleotides located at genome coordinates 8,479-8,707 and 8,864–9,929, respectively. These regions, which correspond to more than 7% of the whole genome, have no known functions. According to Fig. 3, the variability in these regions is not higher than that of other known functional coding regions.
We focused the phylogenetic analysis in the CP considering that is the ORF most largely represented in the coverage map (Fig. 2), and the more relevant viral protein for immunodetection purposes. An alignment of CP ORF fragments was preferred instead of the complete ORF sequences, since the number of sequences available in public databases is 33 instead of 12 (Fig. 2), making the analysis more representative. The most adequate evolution model was the same for CP fragments and for complete CP ORFs [39]. The final alignment considered in the cladogram included 28 non-redundant partial CP ORF sequences. According to Fig. 4, the isolate Cl-817 (EU344894) is excluded from the clade where the rest of the local isolates Cl-664 (EU344895), Cl-766 (EU344893), and Cl-765 (EU 344896) appear close together. Supporting this difference, Cl-817 was the only isolate obtained from the Chilean central region, near Santiago and from a Chardonnay cultivar. The other three isolates were obtained from the VI region, about 150 km south of Santiago and from the Merlot cultivar (Table 2). The same divergence of isolate Cl-817 was obtained when a fragment of all HSP70h available sequences was compared (data not shown). These results are in agreement with the recent work of Prosser et al. [40], who reported highly divergent GLRaV-3 sequences. Dissimilar results have been described by Turturo et al. [41], who reported the presence of single predominant variants of GLRaV-3 in infected grapevines. GLRaV-3 and other closteroviruses infect hosts that persist in the field for a long time and, therefore are exposed to repeated infections. The variability due to inter or intra recombination events that may occur between viruses and hosts in such a long time, is poorly understood [42].

Cladogram of the phylogenetic relationships among partial CP ORFs of different Grapevine leafroll-associated virus-3 isolates. The tree includes the Chilean isolates Cl-664 (EU344895), Cl-766 (EU344893), Cl-765 (EU344896), and Cl-817 (EU344894), which are indicated by the ▲ symbol. An outgroup member ● CPd from Cl-766 isolate was also included. The bootstrap test (1,000 replicates) is shown next to the branches, supporting values higher than 60%. Suffixes represent the country where the isolates were obtained from: It (Italy), Tn (Tunisia), Ch (China), At (Austria), Sy (Syria), Us (USA), Br (Brazil), Ir (Israel), Ng (Nigeria), Gr (Greece), and Cl (Chile)
Although our analysis is limited, it is the first comparison performed of two different complete GLRaV-3 isolates. Before this, the single genome available was not enough for comparative genomic studies. Due to the size and complexity of the GLRaV-3 genome, the limited number of sequences available from different isolates, and the lack of homologous regions on other viruses, several gene products and large intergenic regions remain with unassigned functions. It is possible that future studies on the function of some of the genes will point to the strategy of RNAi silencing, already described in other closteroviruses, such as CTV, GLRaV-2, BYV, and SPCSV [43–46]. Other possible function for the unknown genes of GLRaV-3, may point to the role of humans as carriers of infectious plant viruses and the ability of viral particles to survive in extreme environments like stomach acid pH. It has been recently described that the human gut is a reservoir of several plant viruses, including four grapevine viruses. At least one of the plant viruses described, Pepper mild mottle virus (PMMV), was still infectious in host plants after being isolated from human feces [47].
TAS-ELISA
The TAS-ELISA assay uses a polyclonal antibody to capture the viral particles and a mix of the monoclonals 5A5/C2 and 8G5/H6 as secondary antibodies. These antibodies were produced against the recombinant viral major coat protein of the GLRaV-3 Cl-766 isolate. As a tertiary antibody, a commercial rabbit anti-mouse alkaline phosphatase conjugate was used.
In order to initially determine the performance and titer of the above antibodies, they were individually analyzed as components of a commercial ELISA kit against infected and uninfected grapevine samples. The titer of the capture polyclonal antibody in a DAS- ELISA with a commercial antibody as the secondary conjugate was 1:30,000 (Fig. 5). The monoclonal antibodies 5A5/C2 and 8G5/H6 were analyzed similarly as part of a TAS-ELISA containing commercial capture and tertiary conjugated antibodies. The titer of the monoclonal antibodies was 1:65,000 and 1:16,000, respectively (Fig. 5), which may change depending on the experimental conditions. The TAS-ELISA developed was validated against uninfected and infected grapevines containing single and mixed infections of GLRaV-3, Grapevine fanleaf virus (GFLV), Grapevine leafroll-associated virus-1 (GLRaV-1), Grapevine leafrol-associated virus-2 (GLRaV-2), Grapevine virus A (GVA), and Grapevine fleck virus (GFkV) (Table 2). The test was able to specifically detect GLRaV-3 in the expected samples, which were previously analyzed with RT-PCR and IC-RT-PCR (see below) and no false positives were found. The TAS-ELISA system described in this article is now routinely used in several certification laboratories in Chile and in viral screening in vineyards from the province of Mendoza, Argentina [48].

Titration of polyclonal and monoclonal antibodies using DAS-ELISA and TAS-ELISA against samples from infected (+) and uninfected (−) grapevines. Polyclonal capture antibody was used in a DAS-ELISA combined with a commercial secondary conjugated antibody. Monoclonal antibodies 5A5/C2 and 8G5/H6 were used as secondary antibodies in a TAS-ELISA in combination with a commercial capture antibody and a tertiary rabbit anti-mouse conjugated antibody. Titers were calculated as the antibody dilution corresponding to half the O.D. saturation values
IC-RT-PCR
This assay was developed as a complementary technique for viral detection because of its potentially higher sensitivity. After capturing the viral particles using antibody (same as used for TAS-ELISA) coated microcentrifuge tubes, the viral RNA is released, reverse transcribed, and amplified by PCR in one single tube. The procedure is simple, highly specific, and sensitive [15]. Plants screened with IC-RT-PCR that were infected with GLRaV-3 gave an amplicon of 546 bp corresponding to a fragment of the HSP70h ORF (Fig. 6). The amplified product was cloned and sequenced in each case to confirm its identity. This product was not obtained when the system was applied to grapevine sap containing GLRaV-1, GLRaV-2, GFkV, GVA, and GFLV, in mixed and single infections, thus demonstrating the liability of this technique (Fig. 6, Table 2).

Detection of Grapevine leafroll-associated virus-3 in grapevines by IC-RT-PCR. (A) The virus presence was analyzed in the infected grapevines Cl-766, Cl-765, Cl-664, Cl-817, and in the uninfected grapevine Cl-030. (B) Cross-reaction assay was performed using uninfected and infected grapevines with single and mixed viral infections (virus details in Table 2). “no Ab” corresponds to a control tube without antibody coating, and “pre-I” to a tube coated with pre-immune serum. The amplified product was analyzed by electrophoresis in a 1.5% agarose gel, and sequenced to confirm that it corresponds to a fragment of HSP70h gene of 546 bp (see Table 1)
The approach of producing antibodies from recombinant antigens used in the present work is more convenient and straightforward than the production from purified viral particles because GLRaV-3 cannot be transmitted to herbaceous hosts and, therefore, it is technically difficult to obtain purified virions. Additionally, infected grapevines are likely to contain more than one virus simultaneously [40], making viral purification unpractical and prone to generate non-specific antibodies that may cross-react with other viral species present.
References
G. Herrera, Enfermedades de frutales causadas por virus en Chile. Boletin INIA, (2001), pp. 46–50.
G.P. Martelli, Grapevine virology highlights 2000–2003. in Proceedings of the 14th Meeting of the International Council for the Study of Virus and Virus Diseases of the Grapevine, Locorotondo, Italy, 3–10 (2003).
G.P. Martelli, A.A. Agranovsky, M. Bar-Joseph, D. Boscia, T. Candresse, R.H. Coutts, V.V. Dolja, B.W. Falk, D. Gonsalves, W. Jelkmann, A.V. Karasev, A. Minafra, S. Namba, H.J. Vetten, G.C. Wisler, N. Yoshikawa, Arch. Virol. 147, 2039–2044 (2002). doi:https://doi.org/10.1007/s007050200048
D.G.C. Boscia, P. Gugerli, G.P. Martelli, B. Walter, D. Gonsalves, Vitis 34, 171–175 (1995)
A.V. Karasev, Annu. Rev. Phytopathol. 38, 293–324 (2000). doi:https://doi.org/10.1146/annurev.phyto.38.1.293
C. Cabaleiro, A. Segura, Plant Dis. 81, 283–287 (1997). doi:https://doi.org/10.1094/PDIS.1997.81.3.283
K.S. Ling, H.Y. Zhu, D. Gonsalves, J. Gen. Virol. 85, 2099–2102 (2004). doi:https://doi.org/10.1099/vir.0.80007-0
M.E. Rott, W. Jelkmann, Arch. Virol. 150, 107–123 (2005). doi:https://doi.org/10.1007/s00705-004-0382-z
M. Al Rwahnih, J.K. Uyemoto, B.W. Falk, A. Rowhani, Arch. Virol. 152, 2197–2206 (2007). doi:https://doi.org/10.1007/s00705-007-1064-4
A.V. Karasev, V.P. Boyko, S. Gowda, O.V. Nikolaeva, M.E. Hilf, E.V. Koonin, C.L. Niblett, K. Cline, D.J. Gumpf, R.F. Lee, Virology 208, 511–520 (1995). doi:https://doi.org/10.1006/viro.1995.1182
D.P.C. Jordan, L. Morgan, A. Segaran, Spread of grapevine leafroll and its associated virus in New Zealand vineyards. in Proceedings of the 11th Meeting of the International Council for the Study of Virus and Virus Diseases of the Grapevine, Montreux, Switzerland, 113–114 (1993).
S. Chang, J. Puryear, J. Cairney, Mol. Biol. Rep. 11, 113–116 (1993). doi:https://doi.org/10.1007/BF02670468
M.F. Clark, A.N. Adams, J. Gen. Virol. 34, 475–483 (1977)
D. Zimmermann, M.H. Van Regenmortel, Arch. Virol. 106, 15–22 (1989). doi:https://doi.org/10.1007/BF01311034
K.M. Sefc, W. Leonhardt, H. Steinkellner, J. Virol. Methods 86, 101–106 (2000). doi:https://doi.org/10.1016/S0166-0934(00)00135-X
K.P.J. Rutherford, J. Crook, T. Horsnell, P. Rice, M.A. Rajandream, B. Barrell, Bioinformatics 16, 944–945 (2000). doi:https://doi.org/10.1093/bioinformatics/16.10.944
E.M. Zdobnov, R. Apweiler, Bioinformatics 17, 847–848 (2001). doi:https://doi.org/10.1093/bioinformatics/17.9.847
S.F. Altschul, W. Gish, W. Miller, E.W. Myers, D.J. Lipman, J. Mol. Biol. 215, 403–410 (1990)
C.E. Shannon, Bell Syst. Tech. J. 27, 379–423 & 623–656, (1948)
T.A. Hall, Nucleic Acids Symp. Ser. 41, 95–98 (1999)
J.D. Thompson, D.G. Higgins, T.J. Gibson, Nucleic Acids Res. 22, 4673–4680 (1994). doi:https://doi.org/10.1093/nar/22.22.4673
T.M. Keane, C.J. Creevey, M.M. Pentony, T.J. Naughton, J.O. McLnerney, BMC Evol. Biol. 6, 29 (2006). doi:https://doi.org/10.1186/1471-2148-6-29
G. Schwarz, Ann. Stat. 6(2), 461–464, (1978). doi:https://doi.org/10.1214/aos/117634413610.1214/aos/1176344136
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24, 1596–1599 (2007). doi:https://doi.org/10.1093/molbev/msm092
V. Wilhelm, J. Villegas, A. Miquel, E. Engel, S. Bernales, P.D. Valenzuela, L.O. Burzio, Biol. Res. 36, 223–231 (2003)
J.V. Frangioni, B.G. Neel, Anal. Biochem. 210, 179–187 (1993). doi:https://doi.org/10.1006/abio.1993.1170
U.K. Laemmli, Nature 227, 680–685 (1970). doi:https://doi.org/10.1038/227680a0
A.M. Vaira, M. Vecchiati, V. Masenga, G.P. Accotto, J. Virol. Methods 56, 209–219 (1996). doi:https://doi.org/10.1016/0166-0934(95)01963-4
G. Kohler, C. Milstein, Nature 256, 495–497 (1975). doi:https://doi.org/10.1038/256495a0
N. Fiore, S. Prodan, J. Montealegre, E. Aballay, A.M. Pino, A. Zamorano, J. Plant Pathol. 90, 125–130 (2008)
K.S. Ling, H.Y. Zhu, R.F. Drong, J.L. Slightom, J.R. McFerson, D. Gonsalves, J. Gen. Virol. 79, 1299–1307 (1998)
V.V. Dolja, J.F. Kreuze, J.P. Valkonen, Virus Res. 117, 38–51 (2006). doi:https://doi.org/10.1016/j.virusres.2006.02.002
T.N. Erokhina, R.A. Zinovkin, M.V. Vitushkina, W. Jelkmann, A.A. Agranovsky, J. Gen. Virol. 81, 597–603 (2000)
B. Cevik, R.F. Lee, C.L. Niblett, Arch. Virol. 153, 315–321 (2008). doi:https://doi.org/10.1007/s00705-007-1060-8
T. Satyanarayana, S. Gowda, V.P. Boyko, M.R. Albiach-Marti, M. Mawassi, J. Navas-Castillo, A.V. Karasev, V. Dolja, M.E. Hilf, D.J. Lewandowski, P. Moreno, M. Bar-Joseph, S.M. Garnsey, W.O. Dawson, Proc. Natl. Acad. Sci. U.S.A. 96, 7433–7438 (1999). doi:https://doi.org/10.1073/pnas.96.13.7433
A. Little, C.F. Fazeli, M.A. Rezaian, Virus Res. 80, 109–116 (2001). doi:https://doi.org/10.1016/S0168–1702(01)00343-4
M.S. Bratlie, F. Drablos, BMC Genomics 6, 1 (2005). doi:https://doi.org/10.1186/1471-2164-6-1
D. Raoult, S. Audic, C. Robert, C. Abergel, P. Renesto, H. Ogata, B. La Scola, M. Suzan, J.M. Claverie, Science 306, 1344–1350 (2004). doi:https://doi.org/10.1126/science.1101485
M. Hasegawa, H. Kishino, T. Yano, J. Mol. Evol. 22, 160–174 (1985). doi:https://doi.org/10.1007/BF02101694
S.W. Prosser, D.E. Goszczynski, B. Meng, Virus Res. 124, 151–159 (2007). doi:https://doi.org/10.1016/j.virusres.2006.10.014
C. Turturo, P. Saldarelli, D. Yafeng, M. Digiaro, A. Minafra, V. Savino, G.P. Martelli, J. Gen. Virol. 86, 217–224 (2005). doi:https://doi.org/10.1099/vir.0.80395-0
E. Tanne, I. Sela, Virology 332, 614–622 (2005). doi:https://doi.org/10.1016/j.virol.2004.11.007
M. Chiba, J.C. Reed, A.I. Prokhnevsky, E.J. Chapman, M. Mawassi, E.V. Koonin, J.C. Carrington, V.V. Dolja, Virology 346, 7–14 (2006). doi:https://doi.org/10.1016/j.virol.2005.09.068
R. Lu, A. Folimonov, M. Shintaku, W.X. Li, B.W. Falk, W.O. Dawson, S.W. Ding, Proc. Natl. Acad. Sci. U. S. A. 101, 15742–15747 (2004). doi:https://doi.org/10.1073/pnas.0404940101
K. Ye, D.J. Patel, Structure 13, 1375–1384 (2005). doi:https://doi.org/10.1016/j.str.2005.06.017
J.F. Kreuze, E.I. Savenkov, W. Cuellar, X. Li, J.P. Valkonen, J. Virol. 79, 7227–7238 (2005). doi:https://doi.org/10.1128/JVI.79.11.7227-7238.2005
T. Zhang, M. Breitbart, W.H. Lee, J.Q. Run, C.L. Wei, S.W. Soh, M.L. Hibberd, E.T. Liu, F. Rohwer, Y. Ruan, PLoS Biol. 4, 3 (2006). doi:https://doi.org/10.1371/journal.pbio.0040003
S. Gómez Talquenca, M. Volpe, P. Escobar, E. Engel, O. Gracia, Incidence of three Leafroll Associated Viruses in Cabernet Sauvignon Vineyards of Mendoza, Argentina. in Proceedings of the XI Latin-American Meeting of Enology and Viticulture Mendoza, Argentina, (2007).
Acknowledgments
We thank Professors Nicola Fiore, Patricio Arce, and Juanita Bustamante for plant material, helpful discussions and general support. This research was partially funded by the Chilean Genome Initiative FONDEF G02S1001, UNAB DI-02-06/I, PFB-16/2007, and a Santander-Universia fellowship to EAE.
Author information
Authors and Affiliations
Corresponding author
Additional information
The nucleotide sequence data reported in this article have been submitted to the GenBank nucleotide sequence database and have been assigned the accession numbers EU344893, EU344894, EU344895, and EU344896.
Rights and permissions
About this article
Cite this article
Engel, E.A., Girardi, C., Escobar, P.F. et al. Genome analysis and detection of a Chilean isolate of Grapevine leafroll associated virus-3 . Virus Genes 37, 110–118 (2008). https://doi.org/10.1007/s11262-008-0241-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0241-1