
Abstract
An outbreak of highly pathogenic avian influenza A (H5N1) virus in poultry was reported from Nandurbar and Jalgaon districts of Maharashtra and adjoining areas of Uchhal in Gujarat and Burhanpur in Madhya Pradesh in India from January to April, 2006. In the present study, the full genome of two previously uncharacterized strains of H5N1 viruses isolated at the National Institute of Virology (NIV), Pune, from post-mortem tissues of chicken collected from Navapur, Nandurbar district during the outbreak, has been presented. All the genes belong to clade 2.2 of the Z genotype and are close to the 2006 isolates from Iran, Afghanistan, Mongolia, Italy, and Krasnodar. In a study reported earlier, based on the partial gene sequences of HA, the authors (Pattnaik et al.) hypothesized that the viruses in Jalgaon and Navapur, causing outbreaks 12 days apart, were introduced at different times from different sources. However, our Navapur isolates are closer to the isolate reported from Jalgaon than that from Navapur. Molecular markers suggest that the isolates are sensitive to both drugs Oseltamivir and Amantadine. Amino acid residues responsible for pathogenesis, glycosylation, and receptor binding have also been discussed. The relationship between the Indian viruses and those in the East Africa/West-Asia flyway of migratory birds and the position of Nandurbar in this route suggests that the viruses in India may have been introduced through migratory birds although the role of trade as a possible route of introduction of the virus cannot be ruled out.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Outbreaks of notifiable highly pathogenic avian influenza A (H5N1) viruses in poultry, geese, and other birds or animals have been reported from over 50 countries from Asia, the Middle East, Europe, and Africa. The virus has become endemic in many countries of Central and Southeast Asia.
The H5N1 virus was initially isolated from geese in the Guangdong province of China in 1996 [1]. A series of genetic reassortment events traceable to the precursor of the H5N1 viruses that caused the initial human outbreak in Hong Kong in 1997 and subsequent avian outbreaks in 2001 and 2002 gave rise to a dominant H5N1 genotype (Z) in chickens and ducks that was responsible for the regional outbreaks in 2003–2004 in China and East Asia [2]. Since 2003, the virus has expanded its geographical range to affect poultry in East and Southeast Asia. A new transmission and outbreak wave was initiated after the H5N1 outbreak in migratory waterfowls at Qinghai Lake in May 2005 [3]. The virus further expanded its geographical distribution and caused outbreaks in poultry in over 30 countries in Central Asia, the Middle East, Europe, and Africa [4, 5].
Genetic analysis has confirmed the continued dominance of several lineages of genotype Z viruses in most regions of Asia and genotypes Z, X and G in southern China [6]. A recent study revealed the emergence and predominance of a previously uncharacterized sublineage, “Fujian-like” (FJ-like), in poultry in Southern China since late 2005 [6]. The recognition of multiple sublineages has made it possible to identify the source and to understand the evolutionary and transmission pathways of H5N1 viruses.
In India, the first outbreak of this virus in poultry was notified on February 18, 2006 and seven episodes were recorded from January 27 to April 18, 2006 in the states of Maharashtra, Gujarat, and Madhya Pradesh. No human case was reported. India was declared free of the virus in August 2007. Control measures adopted were stamping out of the entire poultry population including destruction of eggs, feed, litter, and other infected material around a 10 km radius surrounding each outbreak location, restrictions on movement of poultry, poultry products, and disinfection of the infected premises [7].
The major outbreaks in Navapur, Nandurbar (first outbreak), and Jalgaon (second outbreak] districts of Maharashtra occurred within a span of 12 days with heavy mortality in affected chicken populations. Pattnaik et al. [8], based upon partial nucleotide sequences in the HA1 and HA2 regions of the Hemagglutination (HA) gene of one isolate each from both the outbreaks, reported that though both the sequences clustered together with isolates from Iran, Italy, Qinghai, etc. representing subclade 2.2 (Qinghai-like), the virus responsible for the second outbreak had evolved earlier, and both outbreaks were due to different populations of the viruses introduced at two different times. On analysis, the two NA sequences available in the GenBank (accession no. DQ862474, DQ862475) cluster with the sequences from Thailand, Vietnam, and Cambodia suggesting presence of clade 1 of genotype Z virus in Navapur. The occurence of two different types of sequences representing two different subclades, causing poultry outbreaks in the same region within such a short span of time has been intriguing. Therefore, it was considered essential that data on the full genome should be made available to identify the virus strains involved in the outbreaks and to understand the probable source of infection.
In this article we report the complete genome analysis of the two isolates of H5N1 from Navapur, Nandurbar district, Maharashtra, India.
Materials and method
The H5N1 avian influenza outbreaks in India, during January–April, 2006, involved commercial poultry in Navapur, Nandurbar district and backyard poultry in five blocks of Jalgaon district in Maharashtra, commercial poultry in Uchhal, Surat district, Gujarat and Ichhapur, Burhanpur district of Madhya Pradesh. All the affected areas are part of the Tapti River basin and are connected by National and State highways (Fig. 1). The first outbreak was reported in Navapur and Uchhal from January end to mid-February and the second outbreak was in Jalgaon and Ichhapur from February end to mid-April.

Map showing the regions of H5N1 outbreak reports (marked in red) in the states of Maharashtra, Gujarat, and Madhya Pradesh in India
Isolation of virus
Post-mortem chicken tissue samples were collected from two separate farms in Navapur during the first outbreak (early February) in 2006 and stored at −80°C for 8 months before processing. The samples were processed separately according to World Health Organization (WHO) recommendations [9]. Specific-pathogen-free embryonated White Leghorn chicken eggs (SPF eggs) and the Madin Darby Canine Kidney cell line (MDCK) were used for isolation. The isolates were also grown in MDCK without crystalline trypsin [9]. A QuickVue rapid test (Quidel, USA) was carried out with the culture supernatants as an initial rapid diagnostic test. All samples were processed in the enhanced Biosafety level three laboratories.
Identification
Hemagglutination and Hemagglutination inhibition (HAI) tests were performed as described by Kendal et al. [10]. Red blood cells (RBC) from both guinea pig and fowl (0.75% and 0.5% RBC, respectively) were used for the HA test, while only guinea pig RBCs were used for the HAI test. The reference antisera for influenza A/ H5N1, H5N2, H9N2, H7N3, and Newcastle disease virus (NDV) were obtained from the OIE reference laboratory (Venice, Italy).
Viral RNA was extracted using the RNAeasy Viral RNA Mini kit (QIAGEN, Germany). A One-Step reverse transcription-Polymerase Chain Reaction (RT-PCR) (QIAGEN, Germany) was carried out to detect the H5 and N1 specific fragments [11].
Whole genome sequencing
cDNA was prepared from extracted viral RNA using the Uni 12 primer and the eight gene segments (polymerases PB2, PB1, and PA; Hemagglutinin HA, Nucleoprotein NP, Neuraminidase NA, Matrix M, and Nonstructural NS) were amplified using the high fidelity expand HiFi PCR system (Roche, Germany) [12]. PCR products were purified using QIAquick PCR purification and gel extraction kits (QIAGEN, Germany) and sequenced using the Big Dye terminator cycle-sequencing kit (Applied Biosystems, CA, USA) and an automated sequencer (ABI Prism 3130 XL Genetic Analyzer, Applied Biosystems).
Sequence and phylogenetic analyses
Sequences of H5N1 viruses, currently circulating in Asia, Europe and Africa and representative of the various clades of the Z genotype were selected from the Genbank and Los Alamos Influenza Sequence Databases for the phylogenetic analyses. Clades were classified according to the WHO nomenclature [13]. Except for the partial HA sequences of the isolates from Navapur and Jalgaon reported earlier [8] only complete genes were included. For the HA gene, eleven sequences of the 2004-06 isolates from Vietnam, Thailand and Cambodia representing Clade 1, six sequences of the 2004-06 isolates from Indonesia representing Clade 2.1, twenty two sequences of 2005-06 isolates from Asia (Qinghai, Jiangxi, Mongolia, Afghanistan, Iran, India), Europe (Germany, Italy, Turkey), Africa (Nigeria, Egypt, Sudan), Eurasia (Russia-Astrakhan, Krasnoozerka, Novosibirsk, Krasnodar, Kurgan) representing Clade 2.2 and sixteen sequences of the 2004-06 FJ-like and mixed Vietnam isolates [6] representing Clade 2.3 were used. Data sets for the NA gene consisted of five, six, nineteen and nine sequences; for PA nine, six, seventeen and five, for PBI six, four, eighteen and six, for PB2 eight, six, eighteen and six, for NP eight, four, twenty and five, for M ten, five, nineteen and seven and for NS ten, five, twenty and eight sequences representing Clade 1, 2.1, 2.2 and 2.3 respectively, from the same geographical regions were used.
Multiple nucleotide and amino acid sequence alignments for all eight gene segments were performed using Clustalx version 1.83 [14]. MEGA version 3.1 [15] was used for the construction of neighbor-joining phylogenetic trees using the Kimura 2-parameter distance model with 1000 bootstrap replicates. The topology of the trees constructed were confirmed by using the heuristic search maximum likelihood (ML) approach as implemented in PAUP* version 4.0b10 [16] using the F84 nucleotide substitution model and 500 bootstrap replicates. The Guangdong sequences of 1996 and 1997 (clade 3) were used as the outgroup. All strains used for calculations have not been depicted in the trees for the sake of clarity. Glycosylation sites were predicted using the ScanProsite web server [17].
Results
Isolation and identification
One spleen and one trachea sample, from two different farms tested positive for the H5N1 virus. Both isolates showed cytopathic effect (CPE) in the MDCK cell line and killed SPF eggs. The QuickVue rapid test, performed on cell culture supernatants identified these viruses as influenza A/B. The isolates also grew well in MDCK without crystalline trypsin.
In the HAI test, MDCK and egg isolates showed titers of 1:320 and 1:160, respectively, with the influenza H5N1 immune serum and all had a titer of 1:40 with the H5N2 immune serum. There was no inhibition with antisera against H7, H9, and NDV. The H5 and N1 specific primers yielded bands of 219 and 616 bp, respectively, by RT-PCR, suggesting the isolation of H5N1 virus.
Sequence and phylogenetic analysis
The complete genomes of MDCK isolates from the spleen and trachea samples of two different birds, designated A/Ck/India/NIV33487/06 and A/Ck/India/NIV33491/06, respectively (subsequently referred to as 33487 and 33491, respectively), were analyzed in this study. Complete sequences of all eight segments were obtained. The segments PB2, PB1, PA, HA, NP, NA, M, and NS were 2341, 2341, 2233, 1779, 1565, 1398, 1027, and 875 nt, respectively. The GenBank accession numbers of the segments of National Institute of Virology (NIV) isolate 33487 in the same order are EF362425, EF362424, EF362423, EF362418, EF362421, EF362420, EF362419, and EF362422 and of NIV isolate 33491, EF362433, EF362432, EF362431, EF362426, EF362429, EF362428, EF362427, and EF362430. All segments in the two isolates were identical except for one difference (nt 740) in the M gene. The isolates 33487 and 33491 had A and G nucleotides, respectively, resulting in amino acid residues, Tyrosine and Alanine at position 239 in the respective M1 proteins. The HA, NA, and M segments of the egg isolates of the respective viruses were also sequenced and these were identical to the sequences obtained from MDCK isolates.
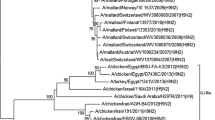
All segments of the NIV isolates clustered within Clade 2.2 of the Z genotype. The percent nucleotide divergence (PND) of the NIV sequences from other members of clade 2.2 ranged from 0.20 in the M2 gene to 0.84 in the HA1 gene and the percent amino acid divergence (PAD) ranged from 0.17 in the PB1 to 1.64 in the NS2 gene (Table 1). The phylogenetic trees obtained for the HA and NA segments are presented in Fig. 2.

Phylogenetic trees constructed by the neighbor-joining method as implemented in MEGA. Nt 28–1734 of the HA gene and nt 20–1369 of the NA gene were used for the analysis. The lengths of the horizontal lines are proportional to the number of nucleotide differences per site. Scale bar indicates number of nucleotide substitutions per site. The trees are rooted at clade 3. Abbreviations: Ck, Chicken; Dk, Duck; Gs, Goose; MDk, Migratory duck; Md, Mallard; Tk, Turkey; BGs, Bar headed goose; Co, Cygnus olor; Cc, Cygnus cygnus; Sw, Swan; Ws, Whooper swan
Hemagglutinin
The NIV isolates were closely related to the isolates from Iran, Mongolia, Krasnodar, Italy, and Afghanistan (Fig 2a). The highest percent nucleotide identity (PNI) was with the isolates from Iran, Italy, and Krasnodar (99.70) and highest percent amino acid identity (PAI) with the isolates from Iran and Italy (99.82). The HA1 sequence showed 99.8 PNI with Iran, Mongolia, and Krasnodar; and 100% amino acid identity with Afghanistan and Mongolia. The HA2 sequence showed 99.7 PNI with the Italy and Krasnodar sequences and 100% amino acid identity with the Iran, Afghanistan, and Italy sequences. The PND of the NIV isolates with other members of clade 2.2 was 0.72 for HA, 0.84 for HA1, and 0.57 for HA2 (Table 1). The PND within clade 2.2 was 0.6 for HA, 0.69 for HA1 and 0.45 for HA2.
The sequences of isolates from India, Afghanistan, Iran, Mongolia, Italy, and Krasnodar had a D154N and a N155D substitutions in the HA1 region while the two 2005 isolates from Qinghai and Jiangxi had only the D154N substitution compared to other members of clade 2.2.
The multibasic amino acid stretch GERRRKKR (single letter code), the marker for high pathogenicity, was present at the cleavage site. Seven potential glycosylation sites, five in HA1 and two in HA2, were identified and mapped on the homology model. The mutation T156A in the NIV strains with respect to Vietnam/04 led to the loss of a potential glycosylation site N154 in HA1 at the 150 loop located on the globular tip of the HA. An additional potential glycosylation site at N543 was noted in HA2.
Of the 14 residues reported to form the receptor-binding domain in HA1 [18], only two mutations K193R and R216K were noted in the NIV isolates. All residues critical for conversion of avian to human receptor specificity, G225D, S227N, G228S, Q226L, S221P, and R216E were avian-type in the NIV isolates. S223A, which is predicted to facilitate binding of sialosides commonly found in mammalian species [13], was not observed in our sequences.
The earlier reported partial sequences from Navapur [8] differ from the NIV sequences by nine amino acids (S163I, N168G, Q169G, E170R, L172P, L173P, V210G, K255Q, and K258Q, wherein in each case the first residue depicts that in the NIV sequences) in the HA1 region (nt 266–856) and two (N465D and L517P) in the HA2 region (nt 1176–1696) and the Jalgaon sequence differs by five amino acids (F143L, Q169G, L172P, T195A and L269M) in HA1 and one (C541S) in the HA2 region. These differences result in a difference of 4.59% and 2.55% with respect to the Navapur sequences and a 1.16% and 0.58% with respect to the Jalgaon sequences for the HA1 and HA2 regions, respectively.
Neuraminidase
The NA phylogenetic tree (Fig. 2b) had a similar topology as the HA tree. The PND within members of clade 2.2 was 0.70. NIV isolates differed from other members of clade 2.2 by 0.64% (Table 1). The highest PNI was with the Iran isolate (99.78) and the highest PAI was with the Iran, Afghanistan, and Qinghai isolates (99.78). There was a unique mutation V247I in the NIV sequences compared to other clade 2.2 isolates. An N228 was observed in the sequences of isolates from Iran, Italy, Krasnodar, Mongolia, Afghanistan, Germany, Nigeria, Astrakhan, and Krasnoozerka while other members of clade 2.2 had S228. N228 was present in the clade 1 NA sequences analyzed.
All five amino acid residues—E119, V149, D151, R156, and E276—responsible for binding the drug Oseltamivir at the active site of neuraminidase [19], were conserved. A homology-based modeled [20, 21] structure of the NA protein built using the 2.5 Ao structure (2HU4) of neuraminidase of A/Vietnam/1203/2004 [19] as the template, showed that the mutation V247I, unique to our isolates, was spatially outside the range of possible non-bonded interactions (data not shown).
The phylogenetic tree (Fig. 2b) showed that the earlier isolates from Navapur clustered with clade 1 sequences. There were 14 differences in amino acids between these and the NIV isolates (3.12 PAD, 2.97 PND).
Polymerases, nucleoprotein, matrix, and nonstructural proteins
The phylogenetic tree topologies of the remaining six gene segments were similar to those observed for HA and NA. The PND/PAD of the NIV isolate with respect to the various clades are depicted in Table 1.
The mutation A239 of the M1 protein of NIV isolate 33487 was unique compared to the other clade 2.2 isolates. The NP protein had two mutations, Y10H and N397S, specific to isolates from India, Afghanistan, Iran, Mongolia, Italy, and Krasnodar in clade 2.2.
Of the 32 host specific amino acid residues [22] in the PB2, PA, NP, M1, and M2 proteins, 31 residues in the NIV isolates indicated avian host specificity. One residue, V28, in the M2 protein suggested human specificity. The amino acid lysine at position 627 associated with pathogenicity in humans is present in our PB2 sequences. The NIV sequences had D and S residues, respectively, in place of N701 and R714 in PB2, which are implicated in enhancing polymerase activity; K and N, respectively, in place of PA N615 and NP K319 that are implicated in moderate enhancement of polymerase activity [23, 24]. One of the two PB1 residues, P13 and N678, which were implicated in dramatic increase in polymerase activity, is present in our sequences (P13) while position 678 has an S [25]. E92 in the NS1 protein, implicated in human adaptation, was present in the NIV isolates [25].
Residues L26, V27, A30, S31, and G34 in the M2 protein, associated with sensitivity to Amantadine [26], were conserved in NIV isolates.
Discussion
All the eight segments of the two NIV isolates belong to clade 2.2 of the Z genotype implying that the viruses belong to the QH-like sublineage, which spread across Asia, Europe, and Africa during 2005–2006. In the HA gene, the clade 2.2 strains that we analyzed clustered into three distinct subgroups, European-Middle Eastern-African (EMA) 1–3, as has been shown recently by Salzberg et al. [27]. This subclustering was verified using the ML method, which resulted in higher bootstrap supports when compared to that observed when using the neighbor-joining method. EMA-1 included 2005 and 2006 isolates from Iraq, Mongolia, Turkey, Egypt, Kurgan, and Novosibirsk. EMA-2 included isolates of 2005 and 2006 from Germany, Nigeria, Astrakhan, and Krasnoozerka, and EMA-3 included the 2006 isolates from India, Afghanistan, Iran, Mongolia, Italy, and Krasnodar. A fourth subgroup consisting of the two 2005 isolates from Qinghai and Jiangxi was also observed, but not assigned any specific name. The EMA-3 subgroup had the D154N and N155D substitutions in the HA1 region while the Qinghai/Jiangxi group had only the D154N substitution when compared to the other subgroups. In the NA gene, the Qinghai strain clustered with EMA-2 subclade. Further, both EMA-2 and EMA-3 sequences had the S228N mutation. It needs to be mentioned that a new unified nomenclature proposed during the Options for the control of Influenza V1 held in Toronto in June 2007, included no further differentiation of clade 2.2 viruses (http://www.who.int/csr/disease/avian_influenza/guidelines/nomenclature/en/index.html). In view of this, the evolution of the emerging patterns within these subclades needs to be studied with time.
An extra glycosylation site at the 150 loop on HA has been associated with high pathogenicity of these viruses for terrestrial domestic poultry, compensating for the reduced virulence conferred by the 20 amino acid deletion in the NA gene [28, 29]. It was also suggested that this glycosylation site being adjacent to the receptor-binding and antigenic sites, is capable of altering the receptor-binding profile and may also help the virus in evading the host antibody response [30]. However, all clade 2.2 viruses analyzed, except the Iran isolate, lack this extra glycosylation site and still retain high pathogenicity in chicken. The receptor-binding domains in our isolates appear to be specific for the avian associated α2–3 linked sialic acid. However, recent reports of a population of ciliated epithelial cells in the human trachea which carry avian receptor-like glycoconjugates at lower densities, and chicken cells which carry human-type sialyl receptors at low concentrations negate a definite species barrier due to receptor specificity [31, 32]. A high virus dose at infection could trigger favorable binding for the development of disease in humans. Indeed, one of the factors thought to be important in the human infections in Indonesia recently is that of exposure to very high doses of the virus.
Residues K627 in PB2, discussed as the single most important residue for mammalian, and in particular, human pathogenicity [33, 34] and P13 in the PB1 gene, associated with increased pathogenicity in mice, are present in the NIV isolates. The combination of D701, R355, and S714 residues in the PB2 gene, found to reduce polymerase activity in mouse experiments [23, 35] was present in our isolates. However, the correlates between mouse pathogenicity assays and human infectivity or disease are still not clear. Of the 33 host specific amino acids in M1, M2, NP, PA, PB2, and NS proteins, the presence of human specific residues, V28 and E92 in M2 and NS1 respectively of the NIV isolates are noteworthy. The significance of the two specific mutations in the NIV isolates, V247I (NA) and A239T (M1), is yet to be ascertained.
Human and animal studies have shown the frequent occurrence of resistance in influenza viruses after exposure to drugs, and drug-resistant viruses transmit without loss of pathogenicity [36–39]. Oseltamivir (Tamiflu) resistant variants have been detected in several influenza A (H5N1) patients [40]. The sequences of NIV isolates indicate sensitivity to the commonly used drugs Amantadine and Oseltamivir. The model of the NA protein indicates no interference in drug binding due to the V247I substitution. The recently reported E248G and Y252H changes in the NA protein, rendering H5N1 isolates about ten times more sensitive to Oseltamivir, were not observed in our isolates [41]. Tamiflu was prescribed to all health care personnel, veterinarians, and investigators working in the outbreak areas, which may be one of the reasons for no human cases in India. A retrospective serology on the samples collected from humans associated with infected poultry, may provide some answers.
The Jalgaon isolate reported earlier was hypothesized to be due to a strain closer to the circulating clade 2.2 viruses and evolved earlier than the Navapur isolate. The sequences of both NIV isolates were closer to other clade 2.2 viruses (Table 1) and to the Jalgaon HA sequence reported than the earlier Navapur HA reported. Also, the NA sequences that we have described belong to clade 2.2 compared to the NA sequences (GenBank accession no. DQ862474, DQ862475) which cluster in clade 1. The source of divergence in the HA region in both the Jalgaon and Navapur isolates reported earlier and the presence of NA of clade 1 needs further study. In view of distinct geographical distributions of strains belonging to clades 1 and 2.2, HA gene in clade 2.2 and NA in clade1 is unlikely. Assuming that the sequence information about the HA and the NA gene of the Navapur/7972 virus is correct, these data are proving a reassortant virus. However, this was not seen in our sequences. Our data suggests presence of the H5N1 virus of clade 2.2 in Navapur also. Sequencing the full genome of other isolates of this outbreak may provide a better understanding.
Large numbers of birds migrate to India every winter by the East Africa/West-Asian, Central Asian and the Black Sea/Mediterranean flyways. The East Africa/West-Asia flyway passes through the epidemic regions in Maharashtra. Distribution of H5N1 viruses to newer areas through migratory birds is an established phenomenon [42]. The close similarity of the NIV isolates to the viruses in the regions of the flyways, strongly suggests that the viruses in India may have been introduced through migratory birds. The role of trade as a possible route of introduction of the virus cannot be ruled out.
The emergence of different lineages and the continued presence of the virus in different parts of the world necessitate monitoring of the virus by regular surveillance in birds and humans and full genome characterization of the isolates to track their movement. The data from such studies would be critical in the initiation and planning of control strategies.
References
X. Xu, K. Subbarao, N.J. Cox, Y. Guo, Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 261, 15–19 (1999)
K.S. Li, Y. Guan, J. Wang et al., Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 430, 209–213 (2004)
H. Chen, G.J.D. Smith, S.Y. Zhang, K. Qin, J. Wang, K.S. Li, et al., Avian flu: H5N1 virus outbreak in migratory waterfowl. Nature 436, 191–192 (2005)
World Organization for Animal Health (2006) Update on avian influenza in animals (Type H5), available at http://www.oie.int/downld/avianinfluenza/A_AI-Asia.htm. Accessed 21 Nov 2006
H. Chen, Y. Li, Z. Li, J. Shi, K. Shinya, G. Deng et al., Properties and dissemination of H5N1 viruses isolated during an influenza outbreak in migratory waterfowl in Western China. J. Virol. 80, 5976–5983 (2006)
G.J. Smith, X.H. Fan, J. Wang, et al., Emergence and predominance of an H5N1 influenza variant in China. Proc. Natl. Acad. Sci. U.S.A. 103, 16936–16941 (2006)
Avian Influenza in India, Follow-up report No. 4 (final report) http://www.oie.int/eng/info/hebdo/AIS_05.HTM#Sec5. Vol. 19, No. 33, Accessed 17 Aug 2006
B. Pattnaik, A.K. Pateriya, R. Khandia, C. Tosh, S. Nagarajan, S. Gounalan, et al., Phylogenetic analysis revealed genetic similarity of the H5N1 avian influenza viruses isolated from HPAI outbreaks in chickens in Maharashtra, India with those isolated from swan in Italy and Iran in 2006. Curr. Sci. 91, 77–81 (2006)
R. Webster, N. Cox, K. Stöhr, Laboratory procedures, in WHO Manual on Animal Influenza Diagnosis and Surveillance, 2nd edn. 15–65 (2002)
A.P. Kendal, M.S. Pereira, J.J. Skehel, Hemagglutinin inhibition. in Concepts and Procedures for Laboratory-Based Influenza Surveillance, ed. by A.P. Kendal, M.S. Pereira, J.J. Skehel, (Centers for Disease Control and Prevention an Pan American Health Organization, Atlanta, 1982), pp. B17–B35
Recommended laboratory tests to identify avian influenza A virus in specimens from humans. WHO June 2005. Available at http://www.who.int/csr/disease/avian_influenza/guidelines/avian-labtests2.pdf. Accessed 21 Nov 2006
E. Hoffmann, J. Stech, Y. Guan, R.G. Webster, D.R. Perez, Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 146, 2275–2289 (2001)
The World Health Organization Global Influenza Program Surveillance Network, Evolution of H5N1 avian influenza viruses in Asia. Emerg. Infect. Dis. 11, 1515–1521 (2005)
D.G. Higgins, J.D. Thompson, T.J. Gibson, Using CLUSTAL for multiple sequence alignments. Meth. Enzymol. 2666, 383–402 (1996)
S. Kumar, K. Tamura, M. Nei, MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5, 150–163 (2004)
D.L. Swofford, PAUP*: Phylogenetic Analysis Using Parsimony (and other methods) 4.0 Beta. (Sinauer Associates, Sunderland, MA, 2002)
E. De Castro, C.J.A. Sigrist, A. Gattiker, V. Bulliard, S. Petra, Langendijk-Genevaux PS et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365 (2006)
J. Stevens, O. Blixt, T.M. Tumpey, J.K. Taubenberger, J.C. Paulson, I.A. Wilson, Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312, 404–412 (2006)
R.J. Russell, L.F. Haire, D.J. Stevens, P.J. Collins, Y.P. Lin, G.M. Blackburn, A.J. Hay, S.J. Gamblin, J.J. Skehel, The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443, 45–49 (2006)
A. Sali, T.L. Blundell, Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993)
N. Guex M.C. Peitsch, SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714 (1997)
M. Shaw L. Cooper, X. Xu et al., Molecular changes associated with the transmission of avian influenza A H5N1 and H9N2 viruses to humans. J. Med. Virol. 66, 107–114 (2002)
G. Gabriel, B. Dauber, T. Wolff, O. Planz, H.D. Klenk, J. Stech, The viral polymerase mediates adaptation of an avian inßuenza virus to a mammalian host. Proc. Natl. Acad. Sci. U.S.A. 102, 18590–18595 (2005)
J.M. Katz, X. Lu, T.M. Tumpey, C.B. Smith, M.W. Shaw, K. Subbarao, Molecular correlates of influenza A H5N1 virus pathogenesis in mice. J. Virol. 74, 10807–10810 (2000)
A.S. Lipatov, S. Andreansky, R.J. Webby, D.J. Hulse, J.E. Rehg, S.S. Krause, et al., Pathogenesis of Hong Kong H5N1 influenza virus NS gene reassortants in mice: the role of cytokines and B- and T-cell responses. J. Gen. Virol. 86, 1121–1130 (2005)
H. Suzuki, R. Saito, H. Masuda, H. Oshitani, M. Sato, I. Sato, Emergence of amantadine-resistant influenza A viruses: epidemiological study. J. Infect. Chemother. 9, 195–200 (2003)
S.L. Salzberg, C. Kingsford, G. Cattoli, D.J. Spiro, D.A. Janies, M.M. Aly, et al., Genome analysis linking recent European and African influenza (H5N1) viruses. Emerg. Infect. Dis. 13(5), 713–718 (2007)
P. Puthavathana, P. Auewarakul, P.C. Charoenying, K. Sangsiriwut, P. Pooruk, K. Boonnak, et al., Molecular characterization of the complete genome of human influenza H5N1 virus isolates from Thailand. J. Gen. Virol. 86, 423–433 (2005)
M.N. Matrosovich, N. Zhou, Y. Kawaoka, R. Webster, The surface glycoproteins of H5 influenza viruses isolated from humans, chicken, and wild aquatic birds have distinguishable properties. J. Virol. 73, 1146–1155 (1999)
H.K. Iwatsuki, R. Kanazawa, S. Sugii, Y. Kawaoka, T. Horimoto, The index influenza A virus subtype H5N1 isolated from a human in 1997 differs in its receptor binding properties from a virulent influenza virus. J. Gen. Virol. 85, 1001–1005 (2004)
M.N. Matrosovich, T.Y. Matrosovich, T. Gray, N.A. Roberts, H.D. Klenk, Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. U.S.A. 101, 4620–4624 (2004)
J.A. Kim, S.Y. Ryu, S.H. Seo, Cells in the respiratory and intestinal tracts of chicken have different proportions of both human and avian influenza virus receptors. J. Microbiol. 43, 366–369 (2005)
O. Werner T.C. Harder, Avian influenza. in Influenza Report 2006, ed. by B.S. Kamps C. Hoffmann W. Preiser (Flying Publisher, Paris, 2006) pp. 48–86
K. Shinya, S. Hamm, M. Hatta, H. Ito, T. Ito, Y. Kawaoka, PB2 amino acid at position 627 affects replicative efficiency, but not cell tropism, of Hong Kong H5N1 influenza A viruses in mice. Virology 320, 258–266 (2004)
Z. Li, H. Chen, P. Jiao, G. Deng, G. Tian, Y. Li et al., Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J. Virol. 79, 12058–12064 (2005)
W.J. Bean, S.C. Threlkeld, R.G. Webster, Biologic potential of amantadine-resistant influenza A virus in an avian model. J. Infect. Dis. 159, 1050–1056 (1989)
G. Boivin, N. Goyette, H. Bernatchez, Prolonged excretion of amantadine-resistant influenza A virus quasi species after cessation of antiviral therapy in an immunocompromised patient. Clin. Infect. Dis. 34, E23–E25 (2002)
C. Sweet F.G. Hayden K.J. Jakeman S. Grambas A.J. Hay, Virulence of rimantadine-resistant human influenza A (H3N2) viruses in ferrets. J. Infect. Dis. 162 969–972 (1991)
F.G. Hayden, R.B. Belshe, R.D. Clover, A.J. Hay, M.G. Oakes, W. Soo, Emergence and apparent transmission of rimantadine-resistant influenza A virus in families. N. Engl. J. Med. 321, 1696–1702 (1989)
WHO inter-country-consultation: influenza A/H5N1 in humans in Asia: Manila, Philippines, http://www.who.int/csr/resources/publications/influenza/WHO_CDS_CSR_GIP_2005_7/en/. Accessed 6–7 May 2005
M.A. Rameix-Welti, F. Agou, P. Buchy, S. Mardy, J.T. Aubin, M.S. Véron et al., Natural variation can significantly alter the sensitivity of influenza A (H5N1) viruses to oseltamivir. Antimicrob. Agents Chemother. 50, 3809–3815 (2006)
B. Olsen, V. Munster, A. Wallensten, J. Waldenström, A.D.M.E. Osterhaus, R.A.M. Fouchier, Global patterns of influenza A virus in wild birds. Science 312, 384–388 (2006)
Acknowledgments
The work described was supported by the Intramural Grant of the National Institute of Virology, Pune (Indian Council of Medical Research, Government of India). The authors wish to thank Dr. A. Chakrabarty, Mr. A. Walimbe, Mr. G. Prabhakar, Mr. N. Mishra, and Mr. S. Ranawade for scientific discussions, bioinformatics analyses and help in the laboratory. We are grateful to Drs. M. Chadhha and D. Mourya for their support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ray, K., Potdar, V.A., Cherian, S.S. et al. Characterization of the complete genome of influenza A (H5N1) virus isolated during the 2006 outbreak in poultry in India. Virus Genes 36, 345–353 (2008). https://doi.org/10.1007/s11262-007-0195-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-007-0195-8