
Abstract
In Korea, extensive Newcastle disease (ND) vaccine programs have been implemented, but ND outbreaks continue to occur occasionally, even in well-vaccinated farms. KBNP-4152 is a virulent ND virus, which has been isolated from vaccinated chickens in Korea. In this study, we conducted a comparison of the antigenicity of KBNP-4152 with that of a vaccine strain, La Sota, via virus-neutralization (VN) and cross haemagglutination-inhibition (HI) tests, and analyzed the genomic sequences. The antigenicity of KBNP-4152 was distinct from La Sota, and the expected genome size was 15,192 nt, as was the case with other recent virulent ND viruses analyzed. Based on the partial F gene, the strain was phylogenetically classified into the VIId genotype, but was distinct from other VII viruses due to amino acid changes at (E347K) and proximal to (M354K), the major linear epitope of HN, as well as relatively low amino acid similarity of the V protein, and a truncated W protein (203 aa vs. 227 aa). Therefore, KBNP-4152 is unique among genotype VII.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Newcastle disease (ND), which is caused by the ND virus (NDV), is a disease that is particularly disastrous in the poultry industry. NDV is an enveloped, single-stranded negative-sense RNA virus, which belongs to the Avulavirus genus of the family Paramyxoviridae [1]. NDV has been classified into three pathotypes, lentogen, mesogen, and velogen types, on the basis of conventional in vivo pathogenicity indices (mean death time of chicken embryo, MDT, and intracerebral pathogenicity index, ICPI) [2]. The NDV genome is comprised of six genes: nucleocapsid protein (NP), phosphoprotein (P), matrix protein (M), fusion protein (F), haemagglutinin-neuraminidase (HN), and large polymerase protein (L) genes [3]. F and HN are present on the envelope. They cooperate in the invasion of host cells, and also determine viral virulence. The multi-basic amino acids at the proteolytic cleavage site in the F protein are relevant to systemic replication, and the HN protein determines the tropisms of NDV [4, 5]. V and W proteins are expressed via RNA editing of the P gene [6]. The V protein functions as an interferon α antagonist by targeting STAT1 for degradation, and V, rather than W, is responsible for the restoration of viral virulence and viral replication [7]. On the basis of phylogenetic analysis with the partial nucleotide sequences of the F genes, NDVs have been classified into 10 genetic groups (I–X) [8–13]. For comparative genomics, several NDV genomes have been sequenced, and three different genome sizes have been defined; 15,186, 15,192, and 15,198 nucleotides (nt) [14–16]. The insertion of six nucleotides into the 5′ non-coding region of the NP gene increased the genome size from 15,186 to 15,192 nt and also the genome size 15,198 was due to a 12 nucleotides insertion in the P gene. The 15,186 and 15,192 nt genomes are shared by genotypes I–IV and V–VIII, respectively [16].
Based on the results of antigenic characterization with monoclonal antibody (MAb), antigenic variants have been reported. However, NDV is still grouped into a serogroup, avian paramyxovirus type-1 (APMV-1) [17, 18]. A single amino acid change, from glutamic acid (E) to lysine (K) at codon 347 (E347K), in the well-characterized, major linear epitope (345PDEQDYQIR353) in HN resulted in the evasion of the antigenic variant from neutralization of mouse MAb [19, 20]. The recognition of the epitope by different host humoral immunities may possibly be different, but the exact same amino acid change was observed in other viruses, including IBA/85 and Korean NDVs [21]. Intensive vaccine programs have been implemented in Korea, but ND outbreaks have occasionally occurred, even in well-vaccinated farms. Therefore, we have characterized the pathoimmunobiological properties of a recent virulent NDV strain, KBNP-4152, and have conducted comparative genomic analysis using the genome nucleotide sequence of KBNP-4152 in order to gain insight into the phenotype–genotype correlation.
Materials and methods
Virus and antiserum
KBNP-4152 was isolated from 42-day-old layer chickens which had been vaccinated three times with live NDV and infectious bronchitis virus (IBV) at 1 (VGGA + H120), 12 (B1 + H120), and 28 (VGGA + H120) days of age, and one time with killed oil vaccine [IBV, NDV (Ulster C), and IBDV; 0.25 ml for each chicken] at 4 days of age, as described previously with slight modification [2]. In brief trachea, cecal tonsil, kidney, and small intestine showing petechiae were homogenized in PBS containing gentamicin (50 μg/ml, Sigma, USA). After centrifugation at 2,000 g for 30 min the supernatant was filtered through a syringe filter (Pall, USA) with 0.2 μm pore size. Around 100 μl of filtered solution was inoculated into 11-day-old specific pathogen-free (SPF) chicken embryos via allantoic cavity. KBNP-4152 was passaged four times through chicken embryo fibroblast (F4) and seven times (E7) through SPF chicken embryo previously. KBNP-4152 (F4E7) and a vaccine strain, La Sota, were propagated in 10 to 11-day-old specific pathogen-free (SPF) chicken embryos, via allantoic cavity inoculation. The harvested allantoic fluids were stored at −70°C until use. The chicken polyclonal antiserum was prepared via the previously described methods [22].
Pathogenicity test
Mean death time (MDT) and intracerebral pathogenicity index (ICPI) were determined as described previously [2].
Cross haemagglutination inhibition (HI) test
The cross HI tests were conducted using chicken polyclonal antisera against KBNP-4152 and La Sota. HI tests were conducted with each virus as described previously [23]. The titer was expressed as a reciprocal of the highest dilution of antiserum, which caused a complete inhibition of agglutination. The antigenic relatedness of KBNP-4152 and La Sota was expressed in R value, as described by Archetti and Horsfall [24].
Virus neutralization (VN) test
The VN titers were determined via the reaction of 2-fold dilutions of anti-La Sota and anti-KBNP-4152 antisera in culture media (199 + F10 media; GIBCO-BRL, NY, USA) with 100-fold quantities of 50% tissue culture infection dose (TCID50) of KBNP-4152 at 37°C for 1 h, after which 25 μl of each virus-serum dilution mixture was inoculated into each well of a 96-well plate containing a monolayer of chicken embryo fibroblast cells for adsorption. After 1 h of adsorption at 37°C, the plates were washed with PBS, and media supplemented with 0.5% FCS was added. After 5 days of incubation, the plates were evaluated for cytopathic effect. The neutralization titer of each serum was expressed as the reciprocal of the highest dilution resulting in a 50% or greater reduction in the number of wells showing viral cytopathic effects. The reproducibility of this assay was determined via the repeated testing of a standard antiserum. The endpoint varied by a ±2-fold dilution.
Isolation of viral RNA, RT-PCR, and sequence and conformational analysis
Nine overlapping amplicons encompassing the majority of the genome of KBNP-4152 were obtained via RT-PCR and LA-PCR, and then cloned into the Topo-TA cloning vector in accordance with the manufacturer’s protocols (Invitrogen, USA). RNA isolation and RT-PCR, and sequence analysis were conducted as previously described [11]. Nucleotide and deduced amino acid sequences were aligned with the Clustal method in the MegAlign program (Window version 3.12e; DNASTAR, Madison, Wis.). Phylogenetic tree was constructed by the neighbor-joining method (Kimura 2-parameter for distance measurement) and the bootstrap test (1,000 repeats) [25]. Bootstrap values lower than 50 were omitted. The computational conformation analysis was performed using Deepview/swiss-pdbViewer V3.7 [26]. All PCR primers used in this study were designed on the basis of the consensus sequences of the genomes of La Sota, ZJ-1, and SF02 and are listed in Table 1.
Accession number
The nucleotide sequence of the viral genome was deposited into the GenBank database, under the accession number DQ839397. The accession numbers of genes used in the sequence analysis are as follows. La Sota (AF077761), ZJ-1 (AF431744), SF02 (AF473851), Ulster (AY562991), Clone 30 (Y18898), B1/47 (AY309418), Beaudette C (M11204, AF064091, AJ225127, X60599, X04687, M24697, M24710, X05399), Herts/33 (AY741404), U.S./Largo/71 (AY562990), IT-227/82 (AJ880277), Guangxi7 (DQ485229), Guangxi9 (DQ485230), Guangxi11 (DQ485231), VGGA (AY289002), SL03 (DQ234579), PX2/03 (DQ520126), Kr-005/00 (AY630423), Kr-188/02(AY630436), ZA378/F/00 (AF532745), ZA331/B/99 (AF532739), JS-5/01(AF456442), Ch/99 (AF358787), TW/98/4 (AF083964), TW/2000 (AF358786), Ch/A7/96 (AY08995), JS-3/00 (AF458010), JS-2/98 (AF456439), JX-2/99 (AF458014), HuB-1/91 (AF378257), D-85/96 (AF001119), Taiwan95 (U62620), MZ46/95 (AF136778), TR8/97 (AF136785), CA/1083/72 (AY562988), Ch/98-1 (AF358785), GB 1168/84 (AF109885), AF2240 (AF048763), JS/1/97 (AF456435), ZhJ-1/85 (AF458023), KJW/49 (AY630409), Aus/32 (M24700), ZJ/1/00/Go (AF456438).
Results
Pathogenicity of KBNP-4152
On the basis of the MDT (45 h) and ICPI (1.99) KBNP-4152 was classified into a velogen-type NDV.
Comparison of antigenicity of KBNP-4152 with a vaccine strain, La Sota
The HI titers of the anti-La Sota antiserum against La Sota and KBNP-4152 were 256 and 32, and those of the anti-KBNP-4152 antiserum were 64 and 256, respectively (Table 2). The R-value was only 18%. The virus neutralization titers of anti-La Sota and anti-KBNP-4152 antisera against KBNP-4152 were 2 and 256, respectively (Table 2).
Phylogenetic analysis
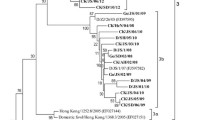
On the basis of the results of phylogenetic analysis with the partial nucleotide sequence of F gene (nt 47–420), KBNP-4152 formed a cluster with Korean, South African, Taiwan, and China viruses, and classified into the VIId genotype (Fig. 1).

Phylogenetic analysis of NDV strains. Numbers below the line indicate the bootstrap values (1,000 replicates). Horizontal distances are proportional to the sequence distances. Phylogenetic tree based on a variable region (nt 47–420) of the fusion (F) protein gene
Analysis of non-coding region of KBNP-4152 viral genome
The genome organization of KBNP-4152 was identical to those of other APMV-1 strains, consisting of six ORFs in the following order: 3′-NP-P-M-F-HN-L-5′. The six-nt insertion in the 5′ non-coding region of the NP gene (between nt 1,647 and 1,648, according to the La Sota strain numbering) was observed in KBNP-4152 as was seen in other strains with 15,192 nt long genomes, and the expected genome size of KBNP-4152 was 15,192. The gene start, gene end, and intergenic region of each of the genes were compared with those of other NDV strains. Those of P and M were conserved in the compared viruses, but those of NP, F, HN, and L were variable. The intergenic region (IGR) sequences between F and HN, and HN and L were relatively variable (Fig. 2). A Uridine insertion and a cytosine deletion were observed in the NP gene end and the IGR of KBNP-4152, just as was observed in other mesogenic and velogenic NDVs. However, a uridine insertion in the gene end of L was observed only in KBNP-4152 among the compared genotype VII viruses (Fig. 2).

Comparison of variable non-coding regions (gene start, gene end, and intergenic sequence) of viral genomic RNA. Homologous nucleotides are represented by dashes
Analysis of coding region of KBNP-4152 viral genome
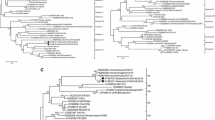
The KBNP-4152 amino acid and nucleotide sequence of NP, P, M, F, HN, and L were compared with those of other NDV strains. All of the KBNP-4152 proteins were found to be most similar to the SF02 isolated from a goose in China, with pair-wise identities in excess of 97.0%, except for P and V. Ulster and La Sota evidenced relatively low similarity to KBNP-4152, SF02, and ZJ-1, with pair-wise amino acid similarities of less than 95.0%. Among the compared proteins, NP and L were relatively conserved, but the P and V proteins were highly variable in the compared NDV strains (Table 3). The V proteins were composed of 239 amino acids and evidenced a variability of between 75.5 and 95.0% among the compared NDV strains. The multi-alignment of the genotype VII V protein amino acids evidenced relatively profound variation (Fig. 3). The lengths of the W proteins varied between 177 and 227 amino acids, and the length of KBNP-4152 was 203 (Table 4). The F protein cleavage site motif of KBNP-4152 was dibasic, 112R-R-Q-K-R-F117, a molecular feature of virulent NDV. The deduced amino acid sequence of the HN protein of KBNP-4152 consisted of 571 amino acids (Table 4). The F and HN protein identities of KBNP-4152 to La Sota were 88.8 and 88.5%, respectively (Table 3). The F and HN amino acid changes, which are relevant to the tertiary structure and charge of proteins, were summarized after comparison with the vaccine strains (Tables 5, 6). No amino acid changes were detected in the F and HN proteins of KBNP-4152, affecting the conformational epitopes. However, interestingly, the primary linear epitope of HN, which consisted of amino acid residues from 345 to 353, evidenced the well-characterized amino acid substitution, E347K (Table 4). One or two amino acid substitutions differing from KBNP-4152 were detected in Largo/71, Guangxi9, Guangxi11, and IT-227/82. Also, an additional amino acid change, M354K, was detected, and we located the changed amino acids on the tertiary structure of HN (Fig. 4). The linear epitope was composed of an α-helix and loop, and was located proximally to the receptor binding pocket. The lysine at 354 was located more proximally than 347K to the receptor binding pocket.

Multi-alignment of amino acid sequences of V protein. Homologous amino acid and stop codon were represented with dot and asterisk, respectively

Location of the HN linear epitope and 354K on the three dimensional structure of HN. The linear epitope from 345 to 353 was represented in yellow except 347K (red color) and 354K was represented in red color. The important residue, 516R, in forming additional receptor binding site (a-RBS) is represented in gray color
Discussion
Although NDVs are generally considered to comprise a single serogroup, their antigenic variations have been detected in several MAb studies [27, 28]. Commercial vaccines protect against mortality [29–32], but the degrees to which they protect against egg drops induced by field NDVs remain questionable. Field NDVs have been commonly isolated from vaccinated chickens, and the existence of antigenic variations conferring lower vaccine immunity have been suspected [10, 28, 33, 34]. The antigenicity of panzootic pigeon APMV-1 during the 1980s and 1990s was distinguished from that of classic viruses via cross HI tests and MAb studies [35]. On the basis of the results of cross HI and VN tests KBNP-4152 was found to be antigenically distinct from La Sota, and the R-value was sufficiently low to corroborate this antigenic dissimilarity. With regard to viral neutralization, F is more important than HN. However, the antigenic variation of HN is greater than that of F [36]. The higher antigenic variability of HN reflects the immunologic pressure and resulting selection of the variant HN. Several MAb studies have revealed several neutralizing antigenic sites within HN [20, 37]. Among them, a linear epitope was reported to consist of amino acids from 345 to 353, and a single amino acid change, E347K, induced the evasion of the mutant virus from MAb neutralization in an experimental model system [18, 19, 38]. The importance of this amino acid change has been ignored thus far, but can be corroborated by the detection of the exact same mutant viruses in the field. Interestingly, prior to 1993, Korean genotype VI viruses possessed 347E, just like all of the commercial vaccine strains. However, SNU9358 (GG), SNU9444, and SNU9598, which were isolated between 1993 and 1995, evidenced the E347K mutation [21]. In addition, identical or somewhat different amino acid changes in the linear epitope were found in the GenBank database (ABB45815, ABB45828, ABB51144, ABB51145, ABB51146, ABB51147, ABB51148, ABB51149, ABB51151, ABB51152, ABB51154, ABC86693, AAY46244, AAY56378, P35742, AY562990, AJ880277, DQ485230, and DQ485231). Therefore, it appears that antigenic variants may have been selected in the field by vaccine immunity. Recently, the three-dimensional (3D) structure of HN has become available [39]. Our computational conformation analysis showed that the linear epitope formed a loop and a partial α-helix, and was located proximally to the receptor binding site (RBS). KBNP-4152 possessed an additional unique amino acid change, M354K, near the linear epitope, and the residue was located more proximally to the RBS than the linear epitope. In addition to its known haemagglutination and neuraminidase activities, HN promotes viral fusion to the host cell membrane, and this activity has been associated with the additional sialic acid binding site (a-RBS) around the 516 residue [40]. The binding of the MAb specific to the linear epitope was shown to inhibit viral infection as well as haemagglutination, and can be explained by the hypothesis that the bound MAb may be positioned above RBS and a-RBS, thereby effecting a blockage of receptor binding and fusion promotion activities via steric hindrance.
The nucleotide sequence of the gene start, the length of the uridine tract of the gene end, and the length of the IGR of the paramyxoviruses affect the efficiency of transcription termination and initiation, and ultimately the gene expression and growth rates, as well as the titer and virulence of the virus [41–43]. The gene start signals of NDVs are conserved among viruses, but differ between the NP/P/M/F/HN (UGCCCAUCUU) and L (UGCCCAUCCU) genes. The lengths of the IGRs between NP-P, P-M, and M-F are one or two nucleotides, but those of F-HN and HN-L are 31 and 47 nucleotides, respectively. The increased length of IGR may be associated with a gradual reduction in the expression from NP to L genes [42, 44]. The gene end signals of the NDVs vary slightly among the viral strains, but that of NP differs between the lentogen (AAUCUUUUUU) and mesogen/velogen (AAUCUUUUUUU) types. The length of the uridine tract of the gene end is relevant to the efficient transcription termination and initiation of downstream genes [43]. If a difference does exist with regard to NP gene transcription termination between the lentogen and mesogen/velogen types, the expression levels of V and W via the RNA editing of the P gene may also differ. Therefore, mesogenic and velogenic viruses may more effectively block the interferon-induced host defense mechanisms. Further studies designed to confirm this hypothesis may prove valuable.
The genome structure of KBNP-4152 is most similar to that of the SF02 strain isolated from a goose in China. The nucleotide identities of each gene of KBNP-4152 and SF02 were less than the amino acid identities in NP, P, M, F, and L, but were identical or higher in P and HN. The former case is common, because of codon redundancy, but the latter is somewhat rare. The substitution of nucleotides is random; however, transition is more frequent than transversion, and the fact that the amino acid identity was less than the nucleotide identity of the P gene can be attributed to natural selection of certain amino acid sequences during viral evolution. The relatively high degree of variability of the P gene has been documented previously, but its biological meaning remains unclear. The P gene encodes for the V and W genes via RNA editing [6]. The NDV V protein, similarly to that of simian virus 5 (SV5), functions as an IFN-α antagonist via the induction of STAT1 degradation [5]. SV5 blocks IFN signaling in human, monkey, and canine cells but fails to block IFN signaling in murine cells. But some amino acid changes within the N-terminal region (NTR) of the SV5 V protein resulted in blocking IFN signaling in murine cells and single amino acid mutation deprived of V protein function [45–47]. Therefore, the NTR of the SV5 V protein is functionally important in the V protein. ND viral strains differ from each other in terms of IFN-inducing capacity and responsiveness to IFNs, and it is noteworthy that the NTR of the ND virus varies among NDVs [48]. STAT2 is required for the degradation of STAT1 by the SV5 V protein, but the amino acid similarity between human and murine STAT2 orthologues is only 68.6%, as compared to 92.4% in STAT1 [49, 50]. Therefore, the V protein of SV5 is functional only in primate STAT2, in which it determines the host restriction of SV5 [51]. NDV induces a more intensive-type 1 IFN response in human cell lines than in chicken embryo fibroblasts, as the V protein determines host restriction [52]. However, differences in V protein activity between waterfowls and fowls, as well as the genetic relatedness of the molecules involved in in-cell immunity, have remained unclear. The STAT1 and STAT2 gene sequences of birds were not available, and the amino acid sequence of IFN-α was compared as a molecular marker to determine the evolutionary distance between waterfowl (goose) and fowl (chicken). Interestingly, the amino acid identity of IFN-α between geese and chickens was only 53%, less than that between humans and mice (62%). Although IFN-α may not be utilized to determine the evolutionary distance of in-cell immunity between waterfowl and fowl, it does appear to be a hint, which extends our knowledge regarding the differences in the virulence of the ND virus and avian influenza virus between waterfowls and fowls. Therefore, considering the unexpected divergence of V and W among ND viruses from the perspective of host adaptation from waterfowl to fowl or vice versa may prove valuable, and may encourage further study in the near future.
In conclusion, KBNP-4152 is unique among genotype VII, and further studies regarding the antigenic characterization of the mutated linear epitope, as well as the biological relevance of the divergent V and W proteins, is required.
References
M.A. Mayo, Arch. Virol. 147, 1071 (2002)
D.J. Alexander, in A Laboratory Manual for the Isolation and Identification of Avian Pathogens, ed. by H.G. Purchase, L.H. Arp, C.H. Domermuth, J.E. Pearson (American Association of Avian Pathologists, Kennett Square, PA, 1989), pp. 114–120
N.S. Millar, P.T. Emmerson, in Newcastle Disease, ed. by D.J. Alexander (Kluwer, Boston, 1988), pp. 79–97
M.S. Collins, J.B. Bashiruddin, D.J. Alexander, Arch. Virol. 128, 363 (1993)
Z. Huang, S. Krishnamurthy, A. Panda, S.K. Samal, J. Virol. 77, 8676 (2003)
M. Steward, I.B. Vipond, N.S. Millar, P.T. Emmerson, J. Gen. Virol. 74, 2539 (1993)
T. Mebatsion, L.T.C. de Vaan, N. de Haas, A. Römer-Oberdörfer, M. Braber, J. Virol. 77, 9259 (2003)
B. Lomniczi, E. Wehmann, J. Herczeg, A. Ballagi-Pordány, E.F. Kaleta, O. Werner, G. Meulemans, P.H. Jorgensen, A.P. Mante, A.L.J. Gielkens, I. Capua, J. Damoser, Arch. Virol. 143, 49 (1998)
J. Herczeg, E. Wehmann, R.R. Bragg, P.M. Travassos Dias, G. Hadjiev, O. Werner, B. Lomniczi, Arch. Virol. 144, 2087 (1999)
C.Y. Yang, H.K. Shieh, Y.L. Lin, P.C. Chang, Avian Dis. 43, 125 (1999)
H.J. Kwon, S.H. Cho, Y.J. Ahn, S.H. Seo, K.S. Choi, S.J. Kim, Vet. Microbiol. 95, 39 (2003)
X.F. Liu, H.Q. Wan, X.X. Ni, Y.T. Wu, W.B. Liu, Arch. Virol. 148, 1387 (2003)
H.J. Tsai, K.H. Chang, C.H. Tseng, K.M. Frost, R.J. Manvell, D.J. Alexander, Vet. Microbiol. 104, 19 (2004)
J. Zou, S. Shan, N. Yao, Z. Gong, Virus Gen. 30, 13 (2005)
A. Czeglédi, D. Ujvári, E. Somogyi, E. Wehmann, O. Werner, B. Lomniczi, Virus Res. 120, 36 (2006)
D. Ujvári, Virus Gen. 32, 49 (2006)
P.H. Russell, D.J. Alexander, Arch. Virol. 75, 243 (1983)
R.M. Iorio, J.B. Borgman, R.L. Glickman, M.A. Bratt, J. Gen. Virol. 67, 1393 (1986)
B. Gotoh, T. Sakaguchi, K. Nishikawa, N.M. Inocencio, M. Hamaguchi, T. Toyoda, Y. Nagai, Virology 163, 174 (1988)
R.M. Iorio, R.J. Syddall, J.P. Sheehan, M.A. Bratt, R.L. Glickman, A.M. Riel, J. Virol. 65, 4999 (1991)
H.J. Kwon, PhD Thesis, Seoul National University (2000)
D.J. Alexander, J.S. Mackenzie, P.H. Russell, Aus. Vet. J. 63, 365 (1986)
D.J. Alexander, R.J. Manvell, P.A. Kemp, G. Parsons, M.S. Collins, S. Brackman, P.H. Russell, S.A. Lister, Avian Pathol. 16, 553 (1987)
I. Archetti, F.L. Horsfall, J. Exp. Med. 92, 441 (1950)
S. Kumar, K. Tamura, M. Nei, Brief. Bioinform. 5, 150 (2004)
N. Guex, M.C. Peitsch, Electrophoresis 18, 2714 (1997)
D.J. Alexander, R.J. Manvell, J.P. Lowings, K.M. Frost, M.S. Collins, P.H. Russell, J.E. Smith, Avian Pathol. 26, 399 (1997)
A. Panshin, E. Shihmanter, Y. Weisman, C. Örvell, M. Lipkind, Comp. Immunol. Microbiol. Infect. Dis. 25, 173 (2002)
D.J. Alexander, G. Campbell, R.J. Manvell, M.S. Collins, G. Parsons, M.S. McNulty, Vet. Rec. 130, 65 (1992)
D.J. Alexander, R.J. Manvell, J. Banks, M.S. Collins, G. Parsons, B. Cox, K.M. Frost, E.C. Speidel, S. Ashman, E.W. Aldous, Avian Pathol. 28, 501 (1999)
P. Roy, A.T. Venugopalan, R. Manvell, Vet. Res. Commun. 24, 135 (2000)
E. Shihmanter, A. Panshin, C. Orvell, Y. Weisman, M. Lipkind, Annual Meeting of the Israel Society of Microbiology, Tel Aviv, 20–23 January, 1997, p. 96
C.Y. Yang, P.C. Chang, J.M. Kwang, H.K. Shieh, Avian Dis. 41, 365 (1997)
L. Yu, Z. Wang, Y. Jiang, L. Chang, J. Kwang, J. Clin. Microbiol. 39, 3512 (2001)
D.J. Alexander, P.H. Russell, M.S. Collins, Vet. Rec. 114, 444 (1984)
T. Morrison, A. Portner, in The Paramyxoviruses, ed. by D.W. Kingsbury (Plenum Press, New York, 1991), pp. 347–482
R.M. Iorio, R.L. Glickman, A.M. Riel, J.P. Sheehan, M.A. Bratt, Virus Res. 13, 245 (1989)
A.C. Samson, M. Nesbit, A.M. Lyon, G. Meulemans, J. Gen. Virol. 69, 473 (1988)
S. Crennell, T. Takimoto, A. Portner, G. Taylor, Nat. Struct. Biol. 7, 1068 (2000)
T.L. Bousse, G. Taylor, S. Krishnamurthy, A. Portner, S.K. Samal, T. Takimoto, J. Virol. 78, 13351 (2004)
A. Kato, K. Kiyotani, M.K. Hasan, T. Shioda, Y. Sakai, T. Yoshida, Y. Nagai, J. Virol. 73, 9237 (1999)
S. Finke, J.H. Cox, K.K. Conzelmann, J. Virol. 74, 7261 (2000)
J.C. Rassa, G.M. Wilson, G.A. Brewer, G.D. Parks, Virology 274, 438 (2000)
M.E. Peeples, in Newcastle Disease, ed. by D.J. Alexander (Kluwer Academic Publishers, Boston, 1988), p. 53
D.F. Young, N. Chatziandreou, B. He, S. Goodbourn, R.A. Lamb, R.E. Randall, J. Virol. 75, 3363 (2001)
N. Chatziandreou, D. Young, J. Andrejeva, S. Goodbourn, R.E. Randall, Virology 293, 234 (2002)
E. Wansley, G.D. Parks, J. Virol. 76, 10109 (2002)
B. Lomniczi, J. Interferon Cytokine Res. 18, 399 (1973)
C. Park, M.J. Lecomte, C. Schindler, Nucleic Acids Res. 27, 4191 (1999)
M. Paulson, S. Pisharody, L. Pan, S. Guadagno, A.L. Mui, D.E. Levy, J. Biol. Chem. 274, 25343 (1999)
J.P. Parisien, J.F. Lau, C.M. Horvath, J. Virol. 76, 6435 (2002)
M.S. Park, A. García-Sastre, J.F. Cros, C.F. Basler, P. Palese, J. Virol. 77, 9522 (2003)
Acknowledgment
This study was supported by the Technology Development Program of the Ministry of Agriculture and Forestry, Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cho, SH., Kim, SJ. & Kwon, HJ. Genomic sequence of an antigenic variant Newcastle disease virus isolated in Korea. Virus Genes 35, 293–302 (2007). https://doi.org/10.1007/s11262-007-0078-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-007-0078-z