
Abstract
Canine Distemper is a highly contagious viral systemic disease that affects a wide variety of terrestrial carnivores. Canine Distemper virus (CDV) appears genetically heterogeneous, markedly in the hemagglutinin protein (H), showing geographic patterns of diversification that are useful to monitor CDV molecular epidemiology. In Mexico the activity of canine distemper remains high in dogs, likely because vaccine prophylaxis coverage in canine population is under the levels required to control effectively the disease. By phylogenetic analysis based on the nucleoprotein (N) and on the H genes, Mexican CDV strains collected between 2007 and 2010 were distinguished into several genovariants, all which constituted a unique group, clearly distinct from field and vaccine strains circulating worldwide, but resembling a CDV strain, 19876, identified in Missouri, USA, 2004, that was genetically unrelated to other North-American CDV strains. Gathering information on the genetic heterogeneity of CDV on a global scale appears pivotal in order to investigate the origin and modalities of introduction of unusual/novel CDV strains, as well as to understand if vaccine breakthroughs or disease epidemics may be somewhat related to genetic/antigenic or biological differences between field and vaccine strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Canine Distemper (CD) is a severe systemic disease caused by a virus classified into the genus Morbillivirus, family Paramyxoviridae, along with Measles virus, Peste des petits ruminants virus, Riderpest virus, Phocine distemper virus and Dolphin morbillivirus (Murphy et al. 1999).
This disease affects a great variety of animals, mainly terrestrial carnivores belonging to the Canidae, Felidae, Mustelidae, Procyonidae, Ursidae and Viverridae families. There are reports on the occurrence of the disease in collared peccaries and Japanese monkeys (Appel et al. 1991; Deem et al. 2000; Yoshikawa et al. 1989). In dogs, the disease is systemic with respiratory, nervous and digestive signs. Footpad hyperkeratosis and enamel hypoplasia have been described and are regarded as characteristic marks of this disease (Appel 1987).
Canine Distemper Virus (CDV) possess a negative sense, single-stranded RNA genome, encoding for 6 main proteins, the Nucleoprotein (N), Phosphoprotein (P), Matrix protein (M), Fusion protein (F), Haemagglutinin (H) and Large protein (L) ( Sidhu et al. 1993). The P gene codes in addition other two proteins, C and V, which have been associated with interferon inhibition and the control of infectivity, respectively (von Messling et al. 2005).
The H protein is involved in SLAM-mediated virus entry in the cells and elicits neutralizing protective antibodies (Jensen et al. 2009). The H gene is the most variable portion of CDV genome and has been used to investigate the genetic relationships among the various strains (Demeter et al. 2007; Haas et al. 1997; Martella et al. 2006). Although being more conserved, the N gene has also been used to gather information on the genetic diversity (Keawcharoen et al. 2005; Yoshida et al. 1998).
On the basis of the full-length sequence of the H gene of CDV strains identified globally, at least seven main geographic groups (genotypes) appear to circulate among the various susceptible hosts, namely America-1 (including the majority of vaccine strains), America-2 (CDVs circulating in Northern America), Asia-1 and Asia-2 (CDVs identified in the Asiatic continent), Europe (viruses of European origin), Arctic (viruses identified in the Arctic ecosystem and in Europe) (Bolt et al. 1997; Iwatsuki et al. 1997; Martella et al. 2007; Pardo et al. 2005). In addition, some CDV strains identified in Africa, Asia and Argentina (Gallo et al. 2007; Woma et al. 2010; Zhao et al. 2010) appear to diverge substantially and might represent separate geographic groups. Besides the vaccines grouped in America-1 genotype there are some independent strains used as vaccines which are related to Rockborn strain (Martella et al. 2011). Nearly 8–9 % aa variation is observable between America-1 CDVs (including the vaccine strains Onderstepoort and Lederle) and the other CDV genotypes in the full-length H protein but the impact of the observed genetic heterogeneity remains unclear.
Despite the availability of CDV vaccines in the market, in Mexico CD remains a major disease of dogs. Limited information is available of the epidemiological and molecular features of the CDV strains circulating locally (Simon-Martínez et al. 2007). In this work we analyzed a total of 14 samples from dogs with clinical signs of Canine Distemper, which were submitted to our laboratories, between the years 2007 and 2010. The viruses were analyzed using two different genomic regions, a fragment of the H gene and N gene.
Materials and methods
Clinical specimens
From 2007 to 2010, 127 samples (whole blood) of animals with suspect of CDV infection were sent to our laboratory for diagnostic confirmation and CDV RNA was identified only in 66 samples (52%) by RT-PCR. Out of 66 samples, 14 could be included in this study based on the amount of the sample and the amount of RNA extracts available for additional analysis. The samples were submitted to our laboratory from private veterinary hospitals in Mexico State (13 samples) and from Jalisco State (1 sample). The clinical signs, age, sex and breed of the patients are summarized in Table 1.
Extraction of nucleic acids
Viral RNA was extracted from leucocytes obtained by washing with nuclease-free water. Briefly, 500 μl of nuclease-free water were added to 500 μl of whole blood in a 1.5 microcentrifuge tube, and centrifuged 7 min at 5,000 g. A total of 500 μl of the supernatant was discarded, then 500 μl of nuclease-free water were added, repeating this step 3 times. Total RNA was extracted with TRI Reagent (Sigma®, USA), following the manufacturer’s instructions.
RT-PCR
A one step RT-PCR was made using the AccessQuick RT-PCR System (Promega®, USA). The final volume of the reaction was 51 μl, and contained 25 μl of AccesQuick Master Mix (2 X), 1 μl of each primer (10 mM), 1 μl of reverse-transcriptase (AMV-RT) (5 u/μl), 18 μl of nuclease-free water and 5 μl of RNA template.
Confirmation of the initial suspect of infection by CDV was accomplished with primers targeted to the N gene, designed by Shin et al. (2004), that amplify a fragment of 297 bp (primer forward 5’-GTAGACGAAGGGTCGAAAG-3’ and primer reverse 5’-GAATCGCCTCAAAGATAGG-3’, nucleotides 507 – 803 of the Onderstepoort strain, AF305419). Thermo-cycling conditions were 45 min at 45°C (reverse-transcription), 5 min at 94°C (initial denaturation), 30 cycles of 30 sec at 94°C, 30 sec at 55°C, 30 sec at 72°C, and a final extension of 10 min at 72°C.
The CDV-positive samples were tested with primers amplifying a 613 bp fragment at the 3’ end of the H gene. The primers were designed in conserved stretches of the H gene (DHIF 5’-TGGTTCACAAGATGGTATTC-3’ and DHIR 5’-CAACACCACTAAATTGGACT-3’). The thermo-cycling condition were as follows: 45 min at 45°C (reverse transcription), 5 min at 94°C (initial denaturation), 30 cycles of 30 sec at 94°C, 30 sec at 51°C, 30 sec at 72°C, and a final extension of 10 min at 72°C.
PCR products sequencing
The amplicons were gel-purified using the Wizard® SV Gel and PCR Clean-Up System (Promega®, USA) and subjected to direct sequencing in a Genetic Analyser 3100 (Applyed Biosystem®, USA) using the Big Dye Terminator v.3.1 Cylce Sequencing Kit (Applyed Biosystem®, USA).
Phylogenetic analysis
The nucleotide sequences were analyzed preliminary with cognate sequences available in GenBank using BLAST software (http://www.ncbi.nlm.nih.gov/). Multiple sequence alignment was made with ClustalW software (Thompson et al. 1994) included in MEGA 4.0 software (Tamura et al. 2007).
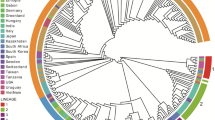
Concatenated sequences of the two gene fragments were aligned and a Neighbor Joining tree was created based on the uncorrected distance matrices, thus identifying the CDV variants circulating in Mexico (Fig. 1).

Phylogenetic tree with concatenated sequences of H and N gene of CDV
For the phylogenetic analysis, the genetic distances were computed using the Kimura two parameter algorithm and the tree was constructed with the Neighbor Joining method with a 1000 bootstrap repeats using the MEGA 4.0 Software (Tamura et al. 2007). Phylogenetic networks were constructed with Network 4.5.1.2 software by the Median Joining algorithm (Bandelt et al. 1999). One Measles strain (AY486084.1) was used as an outgroup in the Neighbor Joining trees.
Results
RT-PCR and sequence analysis
Amplification of fragments of the expected size was obtained with N and H gene and the sequences determined. After removing primer-derived sequences and some sequences with inconsistencies at the 5’ and 3’ ends, a 223 bp-long fragment of the N gene (nucleotides 537 – 759 of the Onderstepoort strain, AF305419) and a 509 bp-long fragment of the H gene (nucleotides 8078 – 8568 of the Onderstepoort strain, GenBank: AF305419) were obtained.
Neighbor Joining analysis with the uncorrected distances generated with the concatenated N/H sequences allowed us to identify at least 10 different genovariants (Fig. 1), EdoMex1(GenBank N gene: FJ490185, H gene: HM771709), EdoMex2 (GenBank N gene: FJ490186, H gene: HM771710), EdoMex4 (GenBank N gene: FJ490188, H gene: HM771711), EdoMex6 (GenBank N gene: FJ490190, H gene: HM771712), EdoMex7 (GenBank N gene: FJ490191, H gene: HM771713), EdoMex8 (GenBank N gene: HM771704, H gene: HM771714), EdoMex9 (GenBank N gene: HM771705, H gene: HM771715), EdoMex10 (GenBank N gene: HM771706, H gene: HM771716), EdoMex11 (GenBank N gene: HM771707, H gene: HM771717), and GDL1 (GenBank N gene: HM771708, H gene: HM771718).
The Mexican genovariants displayed 98.4%-99.9% nucleotide (nt) identity to each other, 91.3%-92.3% nt identity to strain Ondesterpoort and 96.0%-96.6% nt identity to strain A75/17. Deduced amino acid sequence of the H protein fragment was inferred and cysteine and proline amino acids in the Mexican strains were conserved with the majority of CDV strains. Three potential N-glycosilation sites (at position 391–393, 422–424 and 456–458) were found to be conserved as in almost all field CDV strains but strain Onderstepoort that lacks the N-glycosilation sites 391–393 and 456–458.
Phylogenetic analysis
The phylogenetic tree of the N gen grouped all the Mexican variants in the same cluster, distantly related to the vaccine strains and to other field CDV strains identified globally (Fig. 2). Some patterns of segregation were clearly distinguishable (Asian, American and vaccines strains). These segregation patterns were confirmed in the phylogenetic network of the same gen (Fig. 3).

Phylogenetic analysis of N gene of CDV, Neighbor Joining tree. GenBank Accession numbers are indicated in each strain. Bootstrap values lower than 50% are not shown

Phylogenetic analysis of N gene of CDV, Median Joining network
The neighbor joining tree and phylogenetic network generated on the 509 bp-long partial sequences of the H gene (amplified with primers DHIF-DHIR), evidenced geographic patterns (genotypes) resembling the patterns described previously using the full-length H gene (Asia1, Asia2, Europe, Arctic, America1 and America2). All the Mexican CDV variants formed a different group, also including the North American strain 19876, detected in Missouri, USA, 2004 (Pardo et al. 2005) (Figs. 4 and 5).

Phylogenetic analysis of H gene of CDV using Neighbor Joining tree. GenBank Accession numbers are indicated in each strain. Bootstrap values lower than 50% are not shown

Phylogenetic analysis of H gene of CDV, Median Joining network
Discussion
Sequence analysis of CDV strains identified worldwide has been accomplished using different genetic targets, such as the H, F, N or P gene (Bolt et al. 1997; Castilho et al. 2007; Demeter et al. 2007; Haas et al. 1997; Pardo et al. 2005). The use of different genetic targets and different size has generated a large data set of CDV sequences but it has hampered uniform comparison of the various CDV strains. Although the H gene appears to be more suitable for investigating the genetic relationships among CDV strains, haplotypes analysis can also be useful to identify CDV genetic variants (genovariants, i.e. epidemic strains spreading in a given time span or population) and also to identify recombinant viruses.
In this study partial sequences of the H and N genes of CDVs identified from a Mexico region were obtained and compared, using different phylogenetic approaches. Strains displaying 100% nt identity in the N and H sequences were regarded as a unique genovariant. Analysis of 14 CDV strains obtained between 2007–2010, identified 10 distinct genovariants. More genovariants were found to circulate in the same year in the same geographical setting (Edomex 1, 2, 4, 6, 7 in 2007, Edomex 9 and 10 in 2008, Edomex 7 and 11 in 2009). Also, genovariant Edomex7 was found to circulate in 2 distinct years (2007 and 2009) in the same geographical setting.
N gene-based neighbor joining phylogenetic tree and Median Joining network analysis using selected sequences retrieved from GenBank revealed geographic-related patterns of segregation. All the Mexican CDV genovariants were clustered in a distinct group, genetically unrelated to the vaccine CDV strains and to field strains of various geographical origin. A limited number of N gene sequences are available in the GenBank and they span different regions of the gene, thus not being comparable.
The H gene-based neighbor-joining phylogenetic tree and Median Joining network analysis was consistent with similar analysis reported in previous studies (Bolt et al. 1997; Iwatsuki et al. 1997; Martella et al. 2007; Pardo et al. 2005). All the Mexican CDV genovariants clustered in a unique, well-defined (bootstrap value 98%) group. Interestingly, a North American strain, 19876, was found to cluster along with the Mexican viruses. Identity of strain 19876 in the sequenced H gene fragment to the Mexican CDVs was 99.2–99.4 % nt and 99.4–100% aa. Strain 19876 was identified from a dog in Missouri, USA, 2004, and the dog had no history of recent travel outside USA (Pardo et al. 2005). By analysis of the full-length H gene, virus 19876 appeared as the most divergent of the North American CDV strains, as it was found to possess only 93.5–94.6 aa identity to other contemporaneous USA strains (Pardo et al. 2005). Accordingly, given the geographical continuity/proximity, it is safe to assume that 19876-like strains circulate in Mexico and in some geographical areas of USA.
It has been described that changes in N-glycosylation of the H protein may affect neutralization by antibodies and replication in vitro (Harder et al. 1996; Iwatsuki et al. 1997; Lan et al. 2007). On the basis of the partial H protein fragment, the Mexican strains differed from the vaccine strain Ondesterpoort, since they retained N-glycosylation potential sites that are conserved in the majority of field CDVs and that are missing in strain Ondesterpoort.
In conclusion, by phylogenetic analysis of CDV strains, we established a system to distinguish CDV genovariants. We applied this system to investigate the epidemiology of CDV in a Mexican region, individuating several CDV genovariants, highly related to each other, but yet distinguishable. The Mexican genovariants were found to group with a unique North-American CDV strain, genetically unrelated to other North-American viruses. Although the impact of CDV genetic/antigenic heterogeneity is not clear, gathering sequence information on CDVs is useful to understand the epidemiological patterns and global ecology of the virus.
References
Appel MJ (1987) Canine distemper virus. In: Appel MJ (ed) Virus infections of carnivores. Elsevier Science Publishers B.V, Amsterdam, The Netherlands, pp 133–159
Appel MJ, Reggiardo C, Summers BA et al (1991) Canine distemper virus infection and encephalitis in javelinas (collared peccaries). Arch Virol 119:147–152
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 6:37–48
Bolt G, Jensen TD, Gottschalck E et al (1997) Genetic diversity of the attachment (H) protein gene of current isolates of canine distemper virus. J Gen Virol 78:367–372
Castilho JG, Brandão PE, Carnieli JR et al (2007) Molecular analysis of the N gene of canine distemper virus in dogs in Brazil. Arq Bras Med Vet Zoo 59:654–659
Deem SL, Spelman LH, Yates RA, Montali RJ (2000) Canine Distemper in terrestrial carnivores: a review. J Zoo Wildl Med 31:441–451
Demeter Z, Lakatos B, Palade EA et al (2007) Genetic diversity of Hungarian canine distemper virus strains. Vet Microbiol 122:258–269
Gallo MC, Remorini P, Periolo O et al (2007) Detection by RT-PCR and genetic characterization of canine distemper virus from vaccinated and non-vaccinated dogs in Argentina. Vet Microbiol 125:341–349
Haas L, Martens W, Greiser-Wilke I, Mamaev L et al (1997) Analysis of the haemagglutinin gene of current wildtype canine distemper virus isolates from Germany. Virus Res 48:165–171
Harder TC, Kenter M, Vos H et al (1996) Canine distemper virus from diseased large felids: biological properties and phylogenetic relationships. J Gen Virol 77:397–405
Iwatsuki K, Miyashita N, Yoshida E et al (1997) Molecular and phylogenetic analyses of the haemagglutinin (H) proteins of field isolates of canine distemper virus from naturally infected dogs. J Gen Virol 78:373–380
Jensen HT, Nielsen L, Aasted B, Blixenkrone-Moller M (2009) Early life DNA vaccination with the H gene of Canine distemper virus induces robust protection against distemper. Vaccine 27:5178–5183
Keawcharoen J, Theamboonlers A, Jantaradsamee P et al (2005) Nucleotide sequence analysis of nucleocapsid protein gene of canine distemper virus isolates in Thailand. Vet Microbiol 105:137–142
Lan NT, Yamaguchi R, Kawbata A et al (2007) Comparison of molecular and growth properties for two different canine distemper virus clusters, Asia1 and 2, in Japan. J Vet Med Sci 69:739–744
Martella V, Cirone F, Elia G et al (2006) Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy. Vet Microbiol 116:301–309
Martella V, Elia G, Lucente MS et al (2007) Genotyping canine distemper virus (CDV) by a hemi-nested multiplex PCR provides a rapid approach for investigation of CDV outbreaks. Vet Microbiol 16:32–42
Martella V, Blixenkrone-Møller M, Elia G et al (2011) Lights and shades on an historical vaccine canine distemper virus, the Rockborn strain. Vaccine 29(6):1222–1227
Murphy FA, Gibbs EP, Horzinek MC, Studdert MJ (1999) Veterinary virology, 3rd edn. Academic Press, San Diego, CA, pp 411–428
Pardo ID, Johnson GC, Kleiboeker SB (2005) Phylogenetic characterization of canine distemper viruses detected in naturally infected dogs in North America. J Clin Microbiol 43:5009–5017
Shin YJ, Cho KO, Cho HS et al (2004) Comparison of one-step RT-PCR and a nested PCR for the detection of canine distemper virus in clinical samples. Aust Vet J 82:83–86
Sidhu MS, Husar W, Cook SD et al (1993) Canine distemper terminal and intergenic non-protein coding nucleotide sequences: completion of the entire CDV genome sequence. Virology 193:66–72
Simon-Martínez J, Ulloa-Arvizu R, Soriano VE, Fajardo R (2007) Identification of a genetic variant of canine distemper virus from clinical cases in two vaccinated dogs in Mexico. Vet J 175:423–426
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
von Messling V, Svitek N, Cattaneo R (2005) Receptor (SLAM [CD150]) recognition and the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic organs by morbillivirus. J Virol 80:6084–6092
Woma TY, van Vuuren M, Bosman A et al (2010) Phylogenetic analysis of the haemagglutinin gene of current wild-type canine distemper viruses from South Africa: Lineage Africa. Vet Microbiol 143:126–132
Yoshida E, Iwatsuki K, Miyashita N et al (1998) Molecular analysis of the nucleocapsid protein of recent isolates of canine distemper virus in Japan. Vet Microbiol 16:237–244
Yoshikawa Y, Ochikubo F, Matsubara Y et al (1989) Natural infection with canine distemper virus in a Japanese monkey (Macaca fuscata). Vet Microbiol 20:193–205
Zhao J, Yan X, Chai X, Martella V et al (2010) Phylogenetic analysis of the haemagglutinin gene of canine distemper virus strains detected from breeding foxes, raccoon dogs and minks in China. Vet Microbiol 140:34–42
Acknowledgements
The authors would like to thank to the Universidad Autónoma del Estado de México the financial support and to Instituto de Fisiología Celular of the Universidad Nacional Autónoma de México for the sequencing process. The authors also thank Dr. Canio Buonavoglia for the support in the training internship in the Dipartimento di Sanità e Pubblica e Zootecnia, Facoltá di Medicina Veterinaria di Bari. Cesar Gámiz thank to Consejo Nacional de Ciencia y Tecnología for the postgraduated fellowship.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gámiz, C., Martella, V., Ulloa, R. et al. Identification of a new genotype of canine distemper virus circulating in America. Vet Res Commun 35, 381–390 (2011). https://doi.org/10.1007/s11259-011-9486-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-011-9486-6