
Abstract
Expression of the human granulocyte–macrophage colony-stimulating factor (hGM-CSF) gene under the control of the 5′-regulatory sequence of the goat alpha-S1-casein gene with and without a matrix attachment region (MAR) element from the Drosophila histone 1 gene was studied in four and eight transgenic mouse lines, respectively. Of the four transgenic lines carrying the transgene without MAR, three had correct tissues-specific expression of the hGM-CSF gene in the mammary gland only and no signs of cell mosaicism. The concentration of hGM-CSF in the milk of transgenic females varied from 1.9 to 14 μg/ml. One line presented hGM-CSF in the blood serum, indicating ectopic expression. The values of secretion of hGM-CSF in milk of 6 transgenic lines carrying the transgene with MAR varied from 0.05 to 0.7 μg/ml, and two of these did not express hGM-CSF. Three of the four examined animals from lines of this group showed ectopic expression of the hGM-CSF gene, as determined by RT-PCR and immunofluorescence analyses, as well as the presence of hGM-CSF in the blood serum. Mosaic expression of the hGM-CSF gene in mammary epithelial cells was specific to all examined transgenic mice carrying the transgene with MAR but was never observed in the transgenic mice without MAR. The mosaic expression was not dependent on transgene copy number. Thus, the expected “protective or enhancer effect” from the MAR element on the hGM-CSF gene expression was not observed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recombinant protein expression in the milk of transgenic animals has been extensively studied in recent decades (Houdebine 2007, 2009; Bösze and Hiripi 2012). According to this approach, the transgenic animals must be able to synthesize a recombinant human protein and secrete it into milk, where it can then be isolated. This approach has been successfully used in goats, sheep, cows, pigs, and rabbits to produce dozens of human proteins, such as alpha-antitrypsin, serum reactive C protein, antithrombin, blood coagulation factors VIII and IX, lactoferrin, calcitonin, and others (Wall et al. 1997; Lubon 1998; Rudolph 1999; Goldman et al. 2002; Houdebine 2007; Niemann and Kues 2007).
The criteria for correct transgene expression under control of a “milk gene” promoter include the following: (1) the expression must be restricted to the lactating mammary gland only, without ectopic expression in any other tissues; (2) the expression must take place in all of the epithelial cells of the mammary gland, without cell mosaicism; and (3) there must be low variability in transgenic expression between animals. Most researchers believe that incorrect transgene expression and high variability occurs due to random integration of transgenes into the recipient genome. As a result, expression of the transgene depends on the chromatin environment in which it is located, a phenomenon known as the position-effect.
To be expressed correctly, a transgene should contain a promoter, enhancers, insulators, introns, and a transcription terminator (Houdebine 2007; Bösze and Hiripi 2012). A long genomic DNA fragment expressed in bacterial artificial chromosomes or yeast artificial chromosomes often provides correct expression of the transgene (Fujiwara et al. 1997; Rival-Gervier et al. 2002; Tong et al. 2011). However, long DNA fragments can be fragmented during the microinjection procedure (Abrahams et al. 2003; Chandler et al. 2007) or, as suggested by Le Saux et al. (2010), the presence of SINEs or LINEs along the injected DNA can cause multiple rearrangements at the integration site or before integration. For this reason, there is great interest in finding elements that protect transgenes from position-effects.
Scaffold or matrix attachment regions (MARs) are DNA sequences involved in anchoring DNA/chromatin to the nuclear matrix (Harraghy et al. 2008). They are natural components of the eukaryotic genome; therefore, they possess bordering and insulating functions and are able to protect genes from stimulatory and repressive effects arising from flanking genomic regions from neighboring DNA sequences. However, the mechanisms by which MAR elements prevent gene silencing and increase transgene expression are not fully understood (Bode et al. 2000; Girod et al. 2005; Harraghy et al. 2008, 2012). According to computer predictions, there are many thousands of MAR elements in the genome of eukaryotes. However, the testing of protective functions of MAR elements in transgenic animals has been performed with only a few different MARs. For instance, the chicken lysozyme 5′ MAR can confer properly regulated position-independent expression of the whey acidic protein transgene in mice (Bonifer et al. 1990; McKnight et al. 1992, 1996). In addition, this MAR element was able to provide high levels of expression of the hGM-CSF gene but simultaneous low level expression of the human EPO (erythropoietin) gene in transgenic mice (Uusi-Oukari et al. 1997).
In this study, we compared the expression of human GM-CSF under the control of the 5′-regulatory sequence of the goat alpha-S1-casein gene with and without a MAR element in transgenic mice. In a previous study, we demonstrated that a 3,387 bp 5′-flanking sequence of the alpha-S1-casein (CSN1S1) gene from goat was able to provide correct tissue-specific expression of human granulocyte colony-stimulating factor (hG-CSF) in the mammary gland of transgenic mice (Serova et al. 2012). In the current study, we replaced the hG-CSF gene with hGM-CSF and observed correct tissue-specific expression of hGM-CSF in transgenic mice. However, the expression level was somewhat lower than previously reported for hG-CSF. Moreover, we observed ectopic expression of the transgene in one of the four examined transgenic animals. Therefore, we decided to insert a MAR element in this construct to increase the expression of human GM-CSF and prevent ectopic expression. MAR elements taken from one species are typically functional in another species (Kirillov et al. 1996). We chose the MAR element from the Drosophila histone 1 gene described by Mirkovitch et al. (1984) and inserted it upstream of the 5′-regulatory sequence of the goat CSN1S1 gene. It should be noted that this MAR element has not been tested previously on transgenic animals.
We have not found neither enhancer effects of the MAR-element on the expression of hGM-CSF in mammary gland nor its ability to protect the transgene from ectopic expression, but we observed significant cell mosaicism within the mammary gland of the transgenic mice carrying the pMARGoatcasGMCSF transgene.
Materials and methods
Amplification and cloning of the genomic hGM-CSF gene
A full-length gene encoding hGM-CSF was obtained by amplifying human genomic DNA using rTth polymerase (Applied Biosystems, USA) and two primers: GeneU37 (forward) 5′ CCCCGGTCACCATGTGGCTGCAGAGCCTGCTGCTCTT 3′ and GeneL36 (reverse) 5′ TCCCCGCATGCCTGGCCGGTCTCACTCCTGGACTGG 3′. The primers were selected by the published sequence of the human GM-CSF gene (GenBank M13207.1) by means of Oligo6.0. The GeneU37 and GeneL36 primers contain sites for the BstEII and SphI restriction endonucleases, respectively.
A DNA fragment (2,028 bp) representing the hGM-CSF gene was produced by polymerase chain reaction (PCR) using a “GeneAmp®XL PCR Kit” (Applied Biosystems, USA). The PCR reaction mixture consisted of the following: XL PCR buffer, 0.4 μg human genomic DNA, 40 pM forward or reverse primers, 1.2 mM Mg(OAc)2, 4U rTth Polymerase XL, and 200 μM of each dNTP. PCR was performed as follows: an initial denaturation of DNA at 94 °C for 2 min; 10 cycles at 94 °C for 30 s, 70 °C for 40 s with decreasing temperature of 1 °C per cycle, and 68 °C for 2 min; 20 cycles of 94 °C for 30 s, 60 °C for 40 s, and 68 °C for 2 min; and a final cycle at 68 °C for 20 min. The resulting amplified products were separated by electrophoresis in a 1 % agarose gel in Tris–acetate–EDTA (TAE) buffer containing 0.2 μg of ethidium bromide.
The PCR product and the vector plasmid pMGC20 were digested with BstEII and SphI restriction endonucleases, and the digested DNA fragments were then separated by electrophoresis in a 1 % agarose gel using TAE buffer. A DNA fragment containing the hGM-CSF gene and the linearized plasmid pMGC20 were eluted from the agarose gel using a QIAquick Gel Extraction kit (Qiagen, USA) according to the manufacturer’s recommendations. The hGM-CSF gene and the linearized plasmid were then ligated. Cloning procedures were performed according to Sambrook and Russel (2000). To purify the resulting recombinant pMGC-hGM-CSF plasmid, a set of primers completely overlapping the hGM-CSF sequence was used. The plasmids were sequenced with a BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, USA). The reaction products were purified with a DyeEx Spin kit (Qiagen, USA) and separated using an automated ABI 310 genetic analyzer (Applied Biosystems, USA). The nucleotide sequences were analyzed using Sequencher v.4.0.5 software (Gene Codes). Recombinant pMGC-hGM-CSF plasmids containing the full-sized hGM-CSF gene were selected.
Construction of the pGoatcasGMCSF plasmid
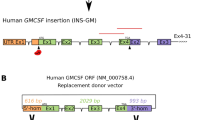
The DNA fragment with the hGM-CSF gene was extracted from the pMGC-hGM-CSF plasmid by BstEII and KpnI hydrolysis. It was then inserted into the pGoatcasGCSF plasmid using the same restriction endonucleases. The pGoatcasGCSF plasmid contained the full-sized human granulocyte colony-stimulating factor (hG-CSF) gene, the 3,387 bp 5′-flanking sequence of the goat alpha-S1-casein (CSN1S1) gene (exon 1, intron 1, and 10 bp of exon 2), as well as the 1,518 bp 3′-flanking sequence of the bovine CSN1S1 gene (exons 18 and 19, and intron 18) (Serova et al. 2012). The hG-CSF gene was replaced by hGM-CSF in the pGoatcasGCSF plasmid. The new recombinant plasmid was called pGoatcasGMCSF (Fig. 1a).

The pGoatcasGMCSF and pMARGoatcasGMCSF expression vectors used to generate and identify transgenic mice, and to express the pGoatcasGMCSF and pMARGoatcasGMCSF transgenes in various tissues of the transgenic mice. a Schematic representation of the pGoatcasGMCSF and pMARGoatcasGMCSF DNA fragments used to generate transgenic mice. Details (from left to right): Drosophila MAR-element; goat CSN1S1 gene 5′-regulatory region; exon 1 (E1), intron 1, and a part of exon 2 (E2) of the goat CSN1S1 gene; a full-sized genomic human GM-CSF gene; exon 18 (E18), intron 18, exon 19 (E19) of a bovine CSN1S1 gene. Additional notations: PrA and PrB—positions for primers used to identify transgenic mice. b Identification of the transgenic mice carrying the pGoatcasGMCSF (left) and pMARGoatcasGMCSF (right) constructs by PCR analysis. Four mice (#3, #9, #11 and #20) that were positive for the pGoatcasGMCSF and eight mice (#M18, #M19, #M20, #M24, #M26, #M28, #M29 and #M43) that were positive for pMARGoatcasGMCSF were identified as transgenic. Non-transgenic mouse DNA was used as a negative control (K−). The DNA used for microinjections served as a positive control (K+). L—100 bp ladder. The PCR product sizes were as predicted (302 bp). c RT-PCR analysis of human GM-CSF and mouse beta-actin transcripts in the mammary glands (1) and organs (2–8) of the transgenic lactating female mice carrying pGoatcasGMCSF (#3, #9, #11, and #20; left) and carrying pMARGoatcasGMCSF (#M18, #M19, #M20, #M24, #M26, M29; right) transgenes. A non-transgenic lactating C57BL female served as a control. 1 mammary gland, 2 liver, 3 lung, 4 spleen, 5 kidney, 6 thymus, 7 salivary gland, and 8 ovary
Construction of the pMARGoatcasGMCSF plasmid
The pMARGoatcasGMCSF plasmid was prepared by addition of the matrix attachment region (MAR) of D. melanogaster upstream of the promoter for the goat CSN1S1 gene in the pGoatcasGMCSF construct. This MAR element contains a specific site for attachment to the chromosome scaffold. The 1,325-bp sequence of the MAR element of D. melanogaster was extracted from GenBank#X14215 (Mirkovitch et al. 1984). Nucleotide sequences from the MAR element were aligned using ClustalX v.1.8 software (Thompson et al. 1997). Primers were designed for amplification of this MAR element by alignment with Oligo6.0 software: MAR_U40 (forward) 5′ GTGTGGTACCGCTAGCATTGACCACGGCCGATTTTAAGTA 3′ and MAR_L37 (reverse) 5′ GAGAGGTACCATTAATGAAATTACATTGATGACGGCA 3′. The primers contained the sequence of the KpnI restriction endonuclease site. A DNA fragment (1,302 bp) containing the MAR element of D. melanogaster was produced by PCR using these primers and the recombinant plasmid pUC-MAR as a template. The recombinant plasmid pUC-MAR, containing the MAR element of D. melanogaster was kindly provided by Dr. Sergey Razin (Institute of Molecular Biology, Moscow, Russia). The resulting amplified products were separated by electrophoresis in a 1 % agarose gel in TAE buffer. Amplicons of the expected size of 1,300-bp were eluted from the agarose gel and purified with a QIAquick Gel Extraction kit (Qiagen, USA).
The pGoatcasGMCSF plasmid and the purified MAR DNA fragment were digested by KpnI restriction endonuclease and then ligated. The resulting recombinant pMARGoatcasGMCSF construct contained the MAR element upstream of the 5′-flanking sequence of the goat CSN1S1 gene (Fig. 1a). Orientation of the MAR insertion was determined by restriction endonuclease analysis and sequencing. The resulting amplified products were separated by electrophoresis in a 1 % agarose gel in TAE buffer containing 0.2 μg of ethidium bromide. Amplicons of the expected size of 1,300-bp were eluted from the agarose gel and purified with a QIAquick Gel Extraction kit (Qiagen, USA). The resulting amplified products were separated by electrophoresis in a 1 % agarose gel in TAE buffer containing 0.2 μg of ethidium bromide. Amplicons of the expected size of 1,300-bp were eluted from the agarose gel and purified with a QIAquick Gel Extraction kit (Qiagen, USA).
Generation of transgenic mice
The pGoatcasGMCSF and pMARGoatcasGMCSF constructs were digested with XbaI/NheI restriction enzymes. Fragments of the digested DNA were separated by electrophoresis in a 0.7 % agarose gel in TAE buffer. A 6,933 bp fragment derived from the pGoatcashGMCSF construct and an 8,237 bp fragment derived from the pMARGoatcasGMCSF construct were eluted from the agarose gel using Qiagen columns (Qiagen, USA,) according to the manufacturer’s recommendations. Purified DNA was dissolved in 0.01 M Tris–HCl–0.25 mM EDTA, pH 7.4. DNA was injected into the zygotes of F1 mice (CBA × C57BL), as described previously (Andreeva and Serova 1992).
Identification of transgenic animals by PCR
Genomic DNA was isolated from the tail tips of 14–20 day old animals, according to http://www.jax.org/imr/tail_nonorg.html. Identification of transgenic animals was accomplished by PCR with the primers: PrA (forward) 5′-AGGAAACTTCCTGTGCAACC-3′; PrB (reverse) 5′-GCTAGACTAGAGGACCAGCAAAA-3′. PrA and PrB are complementary to the 5′-flanking sequence of the goat CSN1S1 gene and the hGMCSF gene, respectively (Fig. 1a). The 25 μl PCR reaction mixture consisted of the following: 0.5–1 μg genomic DNA, 40 nM forward or reverse primers, 0.5 U Taq polymerase, 65 mM Tris–HCl buffer, pH 8.8, containing 16 mM (NH4)2SO4, 0.01 % Tween-20, 2.5 mM MgCl2, and 0.6 mM of each deoxynucleotide (dATP, dCTP, dGTP, and dTTP). PCR was performed as follows: an initial denaturation of DNA at 95 °C for 3 min followed by 28 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 15 s, and extension at 72 °C for 20 s. The resulting amplified products were separated by electrophoresis in a 3 % agarose gel in TAE buffer. The size of the PCR product was 302 bp.
Evaluation of transgene copy number by qPCR
Determination of the pGoatcasGMCSF and pMARGoatcasGMCSF transgene copy number in the transgenic mice by qPCR was performed as described previously (Serova et al. 2012).
Establishment of the chromosomal localization of the pGoatcasGMCSF transgene in transgenic mice by TAIL-PCR
To identify the sites of transgene integration in the mouse genome, Thermal Asymmetric Interlaced PCR (TAIL-PCR) was used (Liu and Chen 2007). Conditions for TAIL-PCR and design of primers were described previously (Serova et al. 2012).
RT-PCR analysis of human GM-CSF gene transcription
The total RNA from various tissues of the transgenic and non-transgenic mice was isolated with TRIzol, converted into cDNA with random primers and the ImProm-II Reverse Transcription System (Promega, USA), and stored at −70 °C (Dvoryanchikov et al. 2005). PCR was performed for the 1 μl of cDNA synthesized in the first RT-PCR as a template in 25 μl of a mixture containing 65 mM Tris–HCl pH 8.8, 16 mM (NH4)2SO4, 0.01 % Tween-20, 2.0 mM MgCl2, 0.2 mM of each deoxynucleotide (dATP, dCTP, dGTP, and dTTP), 0.2 nM of forward and reverse primers, and 1 U of Platinum Taq polymerase. The following primers for human GM-CSF gene transcripts were used: RT-GM-forward 5′-TGCTGCTGAGATGGAATGAAA-3′ and RT-GM-reverse 5′-CCCTTGAGCTTGGTGAGGCT-3′. The reaction was conducted under the following conditions: denaturing for 2 min at 94 °C, 33 cycles (15 s at 94 °C, 15 s at 60 °C and 30 s at 72 °C), and a final 3 min cycle at 72 °C. The expected PCR product was 130 bp in size. As controls, the following primers, complementary to the mouse beta-actin gene, were used: 5′-AGAGGGAAATCGTGCGTGAC-3′ (forward) and 5′-CCAAGAAGGAAGGCTGGAAAA-3′ (reverse). The PCR conditions were as described above for GM-CSF, except for the melting temperature, which was 58 °C. The PCR product for mouse beta-actin was 189 bp in size.
Quantitative immunochemical ELISA analysis of hGM-CSF in milk and blood serum of transgenic mice
Milk samples from the transgenic mice were taken on days 10–12 of lactation. After anesthetization, which was performed 10–20 min prior to the sample collection, 0.5 units of oxytocin was administered to the mice (Uusi-Oukari et al. 1997). The milk samples were frozen and stored at −70 °C. Additionally; samples of blood serum were frozen and stored at −70 °C. The hGM-CSF concentration in milk and serum samples was quantitatively estimated by the immunochemical ELISA method, using monoclonal antibodies against hGM-CSF (R&D Systems, USA) according to the recommendations of the manufacturer. Significance differences between the groups (5–12 animals per group) were assessed by the Mann–Whitney U-test.
Production of rabbit anti-human hGM-CSF polyclonal antibodies
Rabbit immunization was performed according to Harboe et al. (1977). Approximately 100 μg of recombinant hGM-CSF derived from E. coli (kindly provided by Dr. Irina Gileva, NPO Vektor, Novosibirsk, Russia) were loaded on to the SDS-PAGE 13 % gel and ran at a constant voltage of 120 V for 80 min. The gel was stained with Coomassie brilliant blue R250. The fragment containing recombinant hGM-CSF was cut out, destained, homogenized in Freund’s complete adjuvant (Sigma, USA) and subcutaneous injected into the rabbits. Blood was collected 10 days after the last immunization cycle. The immunoglobulin fraction was prepared by adding ammonium sulfate to the blood serum (Brock 1983).
Immobilization of polyclonal antibodies
Polyclonal antibodies were covalently coupled to CNBr-activated Sepharose 4B beads at 5 mg/ml of gel following the manufacturer’s instructions (GE Healthcare). Briefly, the polyclonal antibodies were dialyzed against the coupling buffer (0.1 M NaHCO3 pH 8.3, 0.5 M NaCl) and incubated overnight at 4 °C with CNBr-activated Sepharose 4B. Excess ligand was removed by washing the beads with the coupling buffer and the residual coupling sites on the beads were blocked with 0.1 M Tris–HCl buffer, pH 8.0.
Purification of the hGM-CSF from milk samples of transgenic mice
To isolate the hGM-CSF protein on the affinity columns, we used a commercial chromatography system (ÄKTA purifier 10; Amersham Biosciences) with 10 mM Tris (pH 7.2)/150 mM NaCl (TBS) as the loading buffer and 0.1 M glycine–HCl (pH 2.6) as the elution buffer. Freshly collected mouse milk was centrifuged at 10,000g for 10 min and then cooled to 0 °C to remove the milk fat and insoluble debris. The resulting defatted milk serum was diluted in PBS (1:1 dilution) and loaded onto a polypropylene column (0.8 × 4 cm, Bio-Rad) containing previously immobilized rabbit anti-human hGM-CSF polyclonal antibodies on CNBr-activated Sepharose 4B beads. Elution was performed at a constant flow rate of 0.5 ml/min. Protein fractions of 0.5 ml were collected and the absorbance was monitored at 280 nm. Fractions were immediately neutralized by addition of 2 M Trizma base and concentrated to 20 μl on Centricon-Plus 20 centrifugal filters (Millipore, Bedford, MA).
Immunoblotting of human GM-CSF in the milk of transgenic mice
For immunoblotting, milk samples were fractionated in a 12 % polyacrylamide gel. After electrophoresis, proteins were transferred to nitrocellulose membranes using BioRad equipment in 1× transfer buffer (192 mM glycine, 25 mM Tris). The membranes were then incubated in a blocking solution of 5 % skim milk in PBS for 18 h at 4 °C. After this step, the membranes were incubated for 2 h at room temperature with a primary polyclonal rabbit antibody against hGM-CSF at a concentration of 0.1 mg/ml. Next, the membranes were washed 3 times in 0.1 % Tween 20 in PBS for 10 min. After washing, the membranes were incubated with PBS/Tween containing a secondary goat anti-rabbit IgG/HRP conjugate (Santa Cruz, Biotechnology, USA) for 45 min. Membranes were visualized following incubation with a chemiluminescent cocktail (ECL, Amersham) for 1 min and exposure to Hyperfilm (Amersham). Subsequently, the same membrane was incubated with a solution containing an enzyme substrate (H2O2) and DAB (a chromogenic substance) (0.5 mg/ml diaminobenzidine in 20 mM Tris HCl, pH 7.5, 10 mM imidazole, 0.0006 % H2O2). Samples of recombinant hGM-CSF derived from E. coli with a molecular weight (MW) of approximately 14.5 kDa were used as the non-glycosylated standards.
Immunofluorescent analysis of hGM-CSF in various tissues of transgenic mice
Preparation of frozen specimens and sections of various organs and tissues of transgenic and non-transgenic mice were carried out as described previously (Serova et al. 2012). Immunofluorescent analysis was performed according to the manufacturer’s instructions using a rat monoclonal antibody against hGM-CSF (Santa Cruz Biotechnology, USA) as the primary antibody (diluted 1:100). Goat anti-rat IgG-FITC (Santa Cruz Biotechnology, USA) was used as the secondary antibody (diluted 1:200). After the secondary antibody incubation, the sections were washed three times in PBS with 0.2 % Tween-20, counterstained with 4,6-diamidino-2-phenylindole (DAPI), mounted in a glycerol solution containing 1,4 diazobicyclo-[2.2.3] octane (DABCO; Sigma, USA) and examined by fluorescence microscopy (Axioscope-2; Carl Zeiss, Germany). Images were acquired with WinView software (Roper Scientific Photometrix). Samples of mammary glands and various tissues from at least five transgenic and non-transgenic females of each experimental group were examined.
Estimation of hGM-CSF biological activity in the milk of transgenic mice
Biological activity of hGM-CSF in milk samples was determined from umbilical cord blood as described previously (Serova et al. 2012). Milk samples derived from the transgenic and non-transgenic mice were added to achieve a final hGM-CSF concentration of 10 ng/ml, as determined by ELISA. Cultures were maintained for 12 days in an incubator at 37 °C with 5 % CO2. For morphological identification of cells, the layers of agar were transferred to slides, dehydrated, and stained by May-Grünwald-Giemsa. The GM-CSF activity was expressed in colony-forming units. Significant differences between groups (5–10 animals per group) were assessed by the Mann–Whitney U-test.
Counting of peripheral blood cells
Mice were anesthetized with ether and peripheral blood was collected from their hearts as described previously (Serova et al. 2012). To determine the blood cell differential count, a small drop of the blood was smeared on a clean microscope slide and then stained by Wright–Giemsa. The rest of the blood was analyzed on a hemocytometer (Hemolux 19; Mindray, Shenzhen, China) to determine the number of white blood cells (WBC). Significant differences between groups (4–6 animals per group) were assessed by the Mann–Whitney U-test.
Results
Generation of transgenic mice
A 6,933 bp DNA fragment derived from the pGoatcasGMCSF plasmid and an 8,237 bp DNA fragment derived from the pMARGoatcasGMCSF construct (Fig. 1a) were injected into the pronuclei of mouse zygotes. The injected embryos were transferred into recipients, which in turn gave birth to 65 pups with the pGoatcasGMCSF plasmid and 50 pups with the pMARGoatcasGMCSF plasmid. Using PCR analysis, we identified four females that were positive for the pGoatcasGMCSF. They were designated as #3, #9, #11, and #20 (Fig. 1b). They were mated with non-transgenic males and became founders of four transgenic lines: #3, #9, #11, and #20. Accordingly to the standard nomenclature (Montoliu and Whitelaw 2011), these lines were also named CB/B-F1-Tg(gCsn1s1-hCsf2)3Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)9Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)11Icg and CB/B-F1-Tg(gCsn1s1-hCsf2)20Icg, respectively.
Analysis of the F1 offspring derived from the founders showed that the pGoatcasGMCSF transgene was transmitted to the offspring in a Mendelian fashion. To assess the effect of transgene homozygosity on development and hGM-CSF expression, mice homozygous for the pGoatcasGMCSF transgene were obtained using standard backcrossing between transgenic offspring in each transgenic line.
PCR analysis allowed us to identify 8 animals positive for the pMARGoatcasGMCSF plasmid: #M18, #M19, #M20, #M24, #M26, #M28, #M29, and #M43 (Fig. 1b). Analysis of the F1 offspring obtained from mating the #M18, #M20, #M29, and #M43 founders with non-transgenic mice showed that a 1:1 ratio of transgenic to non-transgenic offspring occurred (i.e., the breeding followed Mendelian inheritance). Transgenic mice were identified in 1 of 12 (a 1:12 ratio), four of 20 (a 1:5 ratio), and two of 12 (a 1:6 ratio) among F1 offspring from the mating of the #M19, #M24, and #M26 founders, respectively, with non-transgenic mice. However, in the next generation derived from these founders, a 1:1 ratio of transgenic to non-transgenic offspring was observed. These data indicate that the integration of the pMARGoatcasGMCSF in the #M18, #M20, #M29, and #M43 founders should have occurred after the 1-cell embryonic stage. Among 24 F1 offspring from the mating of #M28 with non-transgenic mice, we did not observe any transgenic animals. Thus, seven of eight F0 transgenic animals became founders of transgenic lines. Accordingly to the standard nomenclature (Montoliu and Whitelaw 2011) these lines were also named CB/B-F1-Tg(gCsn1s1-hCsf2)M18Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)M19Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)M20Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)M24Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)M26Icg, CB/B-F1-Tg(gCsn1s1-hCsf2)M29Icg and CB/B-F1-Tg(gCsn1s1-hCsf2)M43Icg, respectively.
Determination of copy number and chromosome localization of the pGoatcasGMCSF and pMARGoatcasGMCSF transgenes
Estimation of copy number of the pGoastcasGMCSF and pMARGoatcasGMCSF transgenes was determined by qPCR analysis (Table 1). Table 1 shows that #9, #11, and #20 founders had 2–4 copies of the pGoatcasGMCSF transgene, whereas #GM3 had close to 200 copies per haploid genome. As shown in Table 1, seven founders (#M19, #M20, #M24, #M26, #M28, #M29, and #M43) had 1–3 copies of the pMARGoatcasGMCSF transgene, whereas #M18 had 48 copies of the transgene.
Using TAIL-PCR analysis we determined the 5′-borders of the pGoastcasGMCSF transgene on the chromosomes from all the transgenic lines (Fig. 2). However, we were unable to determine the 3′-borders. A site of transgene integration in the #3 line was identified within the intron of the protein kinase C beta (Prkcb) gene located in region F2 of chromosome 7 (Fig. 2). In the #9 line, the transgene integrated within the intron of the contactin-5 (Cntn5) gene located in region A1 of chromosome 9 (Fig. 2). In lines #11 and #20, the pGoatcasGMCSF transgene was mapped within intergenic regions of E3 on chromosome 9 and C3 on chromosome 6, respectively (Fig. 2). The intragenic integration of the pGoastcasGMCSF transgene into the Prkcb and Cntn5 genes, as well as into intergenic regions of chromosomes 6 and 9, did not accompany any visible developmental abnormalities, as judged from observations of animals homozygous for this transgene.

Chromosomal and genomic localization of the pGoatcasGMCSF transgene in mouse transgenic lines #20, #3, #9, and #11. On the left—regional localization (indicated by arrows) of the transgene on chromosomes 6, 7, and 9 of transgenic mice. On the right—integration sites of the pGoatcasGMCSF transgene on the genomic map at the 5′-transgene-genome junctions in #3, #9, #11, and #20 transgenic mice. Numbers in rectangles reflect positions of the pGoatcasGMCSF transgene in the mouse genome. Cntn5—the contactin-5 gene; Prkc—the protein kinase C beta gene
Unfortunately, we were unable to determine the 5′- and 3′-borders of the pMARGoastcasGMCSF transgene in the seven examined transgenic lines.
Expression of the hGMCSF gene in various tissues of transgenic mice
Figure 1c presents the results of an RT-PCR analysis of the hGCSF transcripts in the mammary glands of lactating transgenic females carrying either the pGoatcasGMCSF or the pMARGoatcasGMCSF transgene. Four transgenic female founders with the pGoatcasGMCSF transgene and six transgenic females with the pMARGoatcasGMCSF contained a 302-bp hGM-CSF transcript that was not present in the non-transgenic females. Beta-actin transcripts were found in all examined females (Fig. 1c). Lactating transgenic females (#M24 and #M43) were negative for the hGM-CSF transcripts.
In three of four transgenic females carrying the pGoatcasGMCSF construct, hGM-CSF transcripts were absent in the liver, lung, spleen and kidney (Fig. 1c). However, lactating female #20 had hGM-CSF transcripts in the lung tissue (Fig. 1c). This finding indicates ectopic expression of the pGoatcasGMCSF transgene in this animal. RT-PCR analysis of various tissues from 6 lactating females with the pMARGoatcasGMCSF demonstrated that two of them (#18 and #19) contained the pMARGoatcasGMCSF transcripts in the lung and salivary gland (Fig. 1c).
Immunochemical ELISA of hGM-CSF in the milk and blood serum of transgenic mice
The results of immunochemical analysis with the use of antibodies against hGM-CSF for transgenic females carrying the pGoatcasGMCSF and the pMARGoatcasGMCSF are given in Table 2. The data show that the concentration of hGM-CSF in the milk of the transgenic females heterozygous for pGoatcasGMCSF varies from 1.9 μg/ml (in #20) to 14 μg/ml (in #11). The animals homozygous for the transgene had hGM-CSF in their milk at levels ranging from 2.7 to 30 μg/ml (Table 2). These data imply that the level of variability in the hGM-CSF concentration in milk can be estimated as moderate.
From the data in Table 2, it follows that the concentration of the hGM-CSF protein in the milk of transgenic mice carrying the pMARGoatcasGMCSF is lower than that in transgenic mice with the pGoatcasGMCSF transgene (Table 2). The values for the pMARGoatcasGMCSF animals ranged from 0.05 μg/ml (in #M24) to 0.7 μg/ml (#M18) (Table 2). As mentioned above, the human GM-CSF transcripts were not found in the mammary gland of transgenic #M28 and #M43 females. Milk from these females did not contain hGM-CSF either.
Table 2 also presents data regarding the amount of hGM-CSF in the blood serum of the transgenic lactating, non-lactating females, and males. hGM-CSF was lacking in the serum of the progeny of founders #3, #9, and #11, but trace amounts (0.1–0.2 ng/ml) were found in the offspring of founder #20 (Table 2). It should be emphasized that hGM-CSF was found in non-lactating transgenic females and males derived from #20. This result is not surprising because ectopic expression of the hGM-CSF was found in the lung of the founder (Fig. 1c).
Immunofluorescent analysis of hGM-CSF in the mammary gland and other tissues of transgenic mice
Figure 3a shows that the mammary gland of a virgin transgenic female lacks cells positive for hGM-CSF. HGM-CSF-positive cells in the mammary gland were identified at day 2 of lactation in females with the pGoatcasGMCSF transgene (Fig. 3b). Most (if not all) secretory epithelial cells of the mammary gland lobules were positive for hGM-CSF in these transgenic females at days 10–15 of lactation, without any signs of cell mosaicism (Fig. 3c). It should be noted that we did not detect cells positive for hGM-CSF in the liver, lung, spleen, thymus, lymph node, kidney, salivary gland, skin, or ovary of transgenic mice derived from founders #3, #9, #11, and #20.

Immunofluorescent analysis of hGM-CSF in various tissues and organs of transgenic females carrying pGoatcasGMCSF or pMARGoatcasGMCSF transgenes, as well as in a non-transgenic female. 1 Mammary gland of a virgin female (#3-217); 2 mammary gland of a female (#20–43) at day 2 of lactation; 3 mammary gland of a female (#9-235) at day 10 of lactation; 4, 5 transgene expression outside the epithelial cells in the mammary gland of #M19-34 (4) and #M20-79 (5) females at day 10 of lactation; 6 Mammary gland of a non-transgenic lactating female; 7, 8 ectopic transgene expression in the salivary gland of lactating #M18-39 (7) and #M19-34 (8) females. Arrows indicate non-epithelial cells positive for hGM-CSF
In contrast, immunofluorescent analysis of hGM-CSF in mammary glands of lactating females with the pMARGoatcasGMCSF transgene demonstrated that hGM-CSF-positive and hGM-CSF-negative secretory epithelial cells were present in various proportions within distinct mammary gland lobules (Fig. 3d, e). Moreover, as follows from Fig. 3d, e, interstitial cells positive for hGM-CSF were detected outside of the lobules. These were presumably adventitial and myoepithelial cells or fibroblasts. This inappropriate expression of the hGM-CSF transgene within can be considered as ectopic expression.
Immunoblotting of human GM-CSF protein in the milk of transgenic mice
Figure 4 presents the results of a western blot of milk samples from transgenic and non-transgenic mice with the use of polyclonal rabbit antibodies against hGM-CSF. As follows from Fig. 4, the recombinant non-glycosylated hGM-CSF derived from E. coli has a MW of 14.5 kDa, whereas hGM-CSF derived from the milk of transgenic mice is represented by at least four bands with MWs between 14.5 and 25 kDa. This pattern is similar or even identical to that of hGM-CSF produced by human phytohemagglutinin-activated lymphocytes (Cebon et al. 1990). Importantly, the non-glycosylated 14.5 kDa from is represented as a minor component in the GM-CSF pattern (Fig. 4) (i.e., hGM-CSF in milk of transgenic mice is presumably represented by several forms with various degrees of glycosylation).

Western blot analysis of hGM-CSF in milk from transgenic and non-transgenic mice. Tg-milk—milk sample from female #11-4, homozygous for the pGoatcasGMCSF transgene. Non-tg milk—negative control, milk sample of a non-transgenic female; K+—positive control is represented by E. coli derived non-glycosylated recombinant hGM-CSF of molecular weight 14.5 kDa. hGM-CSF protein derived from the transgenic female is represented as a set of bands with MWs between 14.5 and 25 kDa, indicating the presence of several forms with various degrees of glycosylation. MW Molecular weight marker
Testing of biological activity of hGM-CSF in milk samples from transgenic mice
To determine the biological activity of hGM-CSF in milk, we examined its effect on precursor cells derived from umbilical cord blood in a colony-generating test. The results presented in Fig. 5 show that the biological activity of hGM-CSF in the milk from transgenic mice is 2–3-fold higher than recombinant bacterial hGM-CSF. Morphological examination of the colonies showed that they were characterized by granulocyte- and monocyte cells at different stages of differentiation. These data suggest a high hematopoietic activity of hGM-CSF in the milk from transgenic mice.

Estimation of biological activity of the hGM-CSF in milk from transgenic and non-transgenic mice by the colony-forming assay. K+ (red)—positive control, E. coli derived non-glycosylated recombinant hGM-CSF; #3, #9, #11 and #20 (green)—milk samples from offspring of transgenic founders; K−—negative control, milk sample from non-transgenic female. The ordinate axis represents the number of colonies formed
Hematological analysis of white blood cells in transgenic mice carrying the pGoatcasGMCSF transgene
Table 3 presents the results of WBC counts in transgenic mice with the pGoatcasGMCSF construct and non-transgenic mice. The data show that there are no differences between transgenic and non-transgenic mice in terms of WBCs including the #20 founder and its offspring positive for hGM-CSF in the blood serum. Additionally, the counts of all types of granulocyte mononuclear cells in transgenic mice are similar to those in non-transgenic animals (Table 3).
Discussion
Our data demonstrate that expression of the hGM-CSF gene directed by a 3,387 bp 5`-flanking sequence of the goat CSN1S1 gene without the MAR element provides expression of the transgene in mammary gland and secretion of the hGM-CSF into the milk in the range of 2–14 μg/ml. In general, our data are comparable with previous results observed in transgenic mice carrying the hG-CSF gene under control of the same promoter (Serova et al. 2012), though the level of hGM-CSF secretion is somewhat lower than that for hG-CSF. There was no correlation between the level of hGM-CSF secretion and the pGoatcasGMCSF transgene copy number. In fact, despite equal copy numbers of the transgene in offspring of #9 and #11 founders, levels of hGM-CSF secretion in these animals differed by more than 10-fold (Table 2). Moreover, offspring from #3, having 200 copies of the pGoatcasGMCSF transgene, did not show high levels of hGM-CSF secretion. However, dose dependence of the pGoatcasGMCSF transgene was observed when comparisons were made of the concentration of hGM-CSF in animals that were homo- and heterozygous for the transgene. Secretion of hGM-CSF was 2-fold higher in homozygous animals than in heterozygous animals for the pGoatcasGMCSF transgene in lines #9, #11, and #20 (low copy number). In line #3 (high copy number), only a 1.5-fold difference was observed.
The 10-fold greater variability in the level of hGM-CSF secretion in #9 and #11 transgenic mice (having similar copy numbers of the transgene) is presumably related to the different integration sites of the transgene in these founders. Both sites of integration of the pGoatcasGMCSF transgene are located in chromosome 9, but in #9 mice, integration occurred within the intron of the Cntn5 gene upstream of the L1 transposable element, whereas in #11 integration occurred within the noncoding region near bp 107, a sequence homologous to mouse leukemia virus (MLV). The cause of the observed variability in the pGoatcasGMCSF transgene expression in #9 and #11 mice is unclear.
Three (#3, #9, and #11) out of the four transgenic mice carrying the hGM-CSF transgene had no signs of ectopic expression outside of the mammary gland, as determined from the RT-PCR, immunochemical, and immunofluorescent analyses. Moreover, it should be noted that there is no cell mosaicism in mammary glands of any transgenic females carrying the hGM-CSF transgene, including the progeny of the founder #9 with pericentromeric localization of this transgene, according to immunofluorescent analysis. Cell mosaicism, manifesting as expression of the transgene in some but not all cells, is a common phenomenon especially for transgenes directed by tissue-specific genes (see Discussion Serova et al. 2012). Some researchers have suggested that pericentromeric localization of transgenes prompts variegated expression (Dobie et al. 1996; Festenstein et al. 1996; Milot et al. 1996), although other studies do not support this notion (Ramirez et al. 2001). Absence of cell mosaicism in the mammary gland with pGoatcasGMCSF transgene expression is in accordance with an earlier study of transgenic mice carrying the human G-CSF gene under the control of the same promoter (Serova et al. 2012).
Presence of hGM-CSF in the blood serum of female and male progeny of founder #20 indicates unambiguous ectopic expression of the transgene. According to RT-PCR analysis, the hGM-CSF transcripts were detected in the lung of #20 lactating females, although hGM-CSF-positive cells in the lung were not revealed by immunofluorescent analysis. This result does not exclude the possibility that certain local cell groups of hGM-CSF-positive cells were missed because the analysis was carried out on small pieces of lung. We were unable to determine a source for the appearance of hGM-CSF in the blood serum of the transgenic line #20. However, immunofluorescent and RT-PCR analyses of liver, lung, spleen, thymus, lymph node, kidney, salivary gland, skin and ovary did not reveal any pGoatcasGMCSF expression. Additionally, the level of hGM-CSF in serum from offspring of founder #20 was very low and did not exert significant influence on hematological indicators of transgenic mice.
Immunoblotting analysis of hGM-CSF in milk from transgenic mice demonstrated that the protein was represented by several forms with MWs from 14.5 to 25 kDa. Similar patterns of hGM-CSF protein in milk from transgenic mice were described previously by Uusi-Oukari et al. (1997). They also demonstrated that hGM-CSF exhibited several forms with varying degrees of glycosylation. Importantly, the biological activity of hGM-CSF in milk samples of all examined transgenic mice was 2–3-fold higher than that of non-glycosylated commercial recombinant GM-CSF derived from E. coli
In general, all expression parameters for the hGM-CSF gene directed by the 3,387 bp 5′-flanking sequence of the goat CSN1S1 gene without the MAR element in transgenic nice resembled those obtained in previous studies of transgenic mice carrying the hG-CSF gene under the control of the same promoter (Serova et al. 2012).
The appearance of a transgenic mouse with ectopic expression of hGM-CSF prompted us to search for elements that might improve construct expression. We decided to insert a MAR element from D. melanogaster into the pGoatcasGMCSF vector upstream of the 3,387 bp 5′-flanking sequence of the goat CSN1S1 gene to prevent negative effects arising from flanking genomic regions neighboring the DNA sequences.
Analysis of eight transgenic mice carrying the pMARGoatcasGMCSF transgene demonstrated the following: (1) the level of secretion of hGM-CSF in milk was lower than that in the transgenic mice carrying the pGoatcasGMCSF construct, and two animals did not express the pMARGoatcasGMCSF transgene; (2) two of the six examined animals showed ectopic expression of the hGM-CSF gene, as indicated by RT-PCR, and hGM-CSF was detected in the blood serum of females and males from three transgenic lines; and (3) mosaic expression of the hGM-CSF gene in epithelial cells of the mammary gland was specific for transgenic mice with the pMARGoatcasGMCSF transgene. Moreover, within the mammary gland, we detected cells that were positive for hGM-CSF outside of the epithelial layer (adventitial and myoepithelial cells and fibroblasts). Mosaic expression was not dependent on the copy number of the pMARGoatcasGMCSF transgene. Thus, we have not observed the expected “protective effect” from the use of a MAR element for improving the expression of the hGM-CSF gene. Interestingly, Kirillov et al. (1996) demonstrated that substitution of the proper MAR of the Ig μ gene with the MAR from Drosophila histone 1 gene did not influence demethylation of the distal promoter. As a result, this produced a high level of expression in the transfected cells.
The most enigmatic phenomenon in our study is the cell mosaicism. Some researchers (Dorer and Henikoff 1994; Garrick et al. 1998) believe that this phenomenon is similar in nature to the well-known position effect observed in D. melanogaster when expression of the targeted gene is influenced by neighboring heterochromatin (Tarlof et al. 1984; Zhimulev 1998). In some cases, integration of the transgene near heterochromatin is accompanied by silencing due to a significant decrease in the proportion of cells that express the transgene (Dobie et al. 1996; Festenstein et al. 1996). However, cell mosaicism was observed in transgenic mice with transgene localization at pericentromeric and outside regions (Ramirez et al. 2001). For instance, the chicken lysozyme 5′ MAR element fused with the keratin 5 promoter did not prevent cell mosaicism in stratified epithelia of mice with transgene localizations at pericentromeric and outside regions (Ramirez et al. 2001).
Additionally, increased copy number of the transgene can induce silencing, which in turn, is frequently manifested as a decrease in the proportion of cells that express the transgene, resulting in a variegated pattern of expression. In contrast, reduction in the transgene copy number results in a marked increase in expression of the transgene and simultaneous increase in the number of cells with the active transgene (Garrick et al. 1998). However, we did not find any difference in degree of mosaicism among epithelial cells from mammary glands in #M18 and #M19 mice carrying either 48 copies or 1 copy of the pMARGoatcasGMCSF transgene, respectively.
Taken together, we have not observed the expected “protective or enhancer effect” from the use of the MAR element from the Drosophila histone 1 gene for improving the expression of the hGM-CSF gene but the used MAR was able to reinforce cell mosaicism.
References
Abrahams BS, Chong ACO, Nisha M, Milette D, Brewster DA, Berry ML, Muratkhodjaev F, Mai S, Rajcan-Separovic E, Simpson EM (2003) Metaphase FISHing of transgenic mice recommended: FISH and SKY define BAC-mediated balanced translocation. Genesis 36:134–141
Andreeva LE, Serova IA (1992) Effect of micromanipulations used for transgenesis on the early development of mice. Ontogenez (Russian) 23:637–643
Bode J, Benham C, Knopp A, Mielke C (2000) Transcriptional augmentation: modulation of gene expression by scaffold/matrix attached regions (S/MAR elements). Crit Rev Eukaryot Gene Expr 10:73–90
Bonifer C, Vidal M, Grosveld F, Sippel AE (1990) Tissue specific and position independent expression of the complete gene domain for chicken lysozyme in transgenic mice. EMBO J 9:2843–2848
Bösze Z, Hiripi L (2012) Recombinant protein expression in milk of livestock species. Methods Mol Biol 824:629–641
Brock TD (1983) Membrane filtration: a user’s guide and reference manual. Science Tech, Madison, WI
Cebon J, Nicola N, Ward M, Gardner I, Dempsey P, Layton J, Dührsen U, Burgess AW, Nice E, Morstyn G (1990) Granulocyte-macrophage colony stimulating factor from human lymphocytes. The effect of glycosylation on receptor binding and biological activity. J Biol Chem 265:4483–4491
Chandler KJ, Chandler RL, Broeckelmann EM, Hou Y, Southard-Smith EM, Mortlock DP (2007) Relevance of BAC transgene copy number in mice: transgene copy number variation across multiple transgenic lines and correlations with transgene integrity and expression. Mamm Genome 18:693–708
Dobie KW, Lee M, Fantes JA, Graham E, Clark AJ, Springbett A, Lathe R, McClenagham M (1996) Variegated transgene expression in mouse mammary gland is determined by the transgene integration locus. Proc Natl Acad Sci USA 93(6659–6664):827
Dorer DR, Henikoff S (1994) Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 77:1–20
Dvoryanchikov GA, Serova IA, Andreeva LE, Dias LPB, Azevedo S, Serov OL (2005) Secretion of biologically active human granulocyte colony-stimulating factor (G-CSF) in milk of transgenic mice. Russ J Genet 41:1088–1094
Festenstein R, Tolaini M, Corbella P, Mamalaki C, Parrinton J, Fox M, Miliou A, Jones M, Kioussis D (1996) Locus control region function and heterochromatin-induced position effect variegation. Science 271:1123–1125
Fujiwara Y, Miwa M, Takahashi R, Hirabayashi M, Suzuki T, Ueda M (1997) Position-independent and high-level expression of human alpha-lactalbumin in the milk of transgenic rats carrying a 210-kb YAC DNA. Mol Reprod Dev 47:157–163
Garrick D, Fiering S, Martin DIK, Whitelaw E (1998) Repeat-induced gene silencing in mammals. Nat Genet 18:56–59
Girod PA, Zahn-Zabal M, Mermod N (2005) Use of the chicken lysozyme 5 matrix attached region to generate high producer CHO cell lines. Biotechnol Bioeng 91:1–11
Goldman IL, Kadulin SG, Razin SV (2002) Transgenic goats in the world’s pharmaceutical industry in the XXI century. Russ J Genet 38:1–14
Harboe M, Closs O, Bjorvatn B, Kronvall G, Axelsen NH (1977) Antibody response in rabbits to immunization with Mycobacterium leprae. Infect Immun 18:792–805
Harraghy N, Gaussin A, Mermod N (2008) Sustained transgene expression using MAR elements. Curr Gene Ther 8:353–366
Harraghy N, Buceta M, Regamey A, Girod PA, Mermod N (2012) Using matrix attachment regions to improve recombinant protein production. Methods Mol Biol 801:93–110
Houdebine LM (2007) Transgenic animal models in biomedical research. Methods Mol Biol 360:163–202
Houdebine LM (2009) Production of pharmaceutical proteins by transgenic animals. Comp Immunol Microbiol Infect Dis 32:107–121
Kirillov A, Kisler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y (1996) A role for nuclear NF-κB in B-cell-specific demethylation of the Igκ locus. Nat Genet 13:435–441
Le Saux A, Houdebine L-M, Jolivet G (2010) Chromosome integration of BAC (bacterial artificial chromosome): evidence of multiple rearrangements. Transgenic Res 19:923–931
Liu Y-G, Chen Y (2007) High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 43:649–656
Lubon H (1998) Transgenic animal bioreactors in biotechnology and production of blood proteins. Biotechnol Annu Rev 4:1–54
McKnight RA, Shamay A, Sankaran L, Wall RJ, Hennighausen L (1992) Matrix-attachment regions can impart position-independent regulation of a tissue-specific gene in transgenic mice. Proc Natl Acad Sci USA 89:6943–6947
McKnight RA, Spencer M, Wall RJ, Hennighausen L (1996) Severe position effects imposed on a 1 kb mouse whey acidic protein gene promoter are overcome by heterologous matrix attachment regions. Mol Reprod Dev 44:179–184
Milot E, Strouboulis J, Trimborn T, Wijgerde M, De Boer E, Langeveld A, Tan-Un K, Vergeer W, Yannoutsos N, Grosveld F, Fraser P (1996) Heterochromatin effects on the frequency and duration of LCR-mediated gene transcription. Cell 87:105–114
Mirkovitch J, Mirault M-E, Laemmle UK (1984) Organization of the higher-order chromatin loop: specific DNA attachment sites on nuclear scaffold. Cell 39:223–232
Montoliu L, Whitelaw CBA (2011) Using standard nomenclature to adequately name transgenes, knockout gene alleles and any mutation associated to a genetically modified mouse strain. Transgenic Res 20:435–440
Niemann H, Kues WA (2007) Transgenic farm animals: an update. Reprod Fertil Dev 19:762–770
Ramirez A, Milot E, Ponsa I, Marcos-Gutiérrez C, Page A, Santos M, José Jorcano J, Miguel Vidal M (2001) Sequence and chromosomal context effects on variegated expression of keratin 5/lacZ constructs in stratified epithelia of transgenic mice. Genetics 158:341–350
Rival-Gervier S, Viglietta C, Maeder C, Attal J, Houdebine LM (2002) Position-independent and tissue-specific expression of porcine whey acidic protein gene from a bacterial artificial chromosome in transgenic mice. Mol Reprod Dev 63:161–167
Rudolph NS (1999) Biopharmaceutical production in transgenic livestock. Trends Biotechnol 17:367–374
Sambrook J, Russel DW (2000) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Serova IA, Dvoryanchikov GA, Andreeva LE, Burkov IA, Dias LPB, Battulin NR, Smirnov AV, Serov OL (2012) A 3,387 bp 50-flanking sequence of the goat alpha-S1-casein gene provides correct tissue-specific expression of human granulocyte colony-stimulating factor (hG-CSF) in the mammary gland of transgenic mice. Transgenic Res 21:485–498
Tarlof KD, Hobbs C, Jones H (1984) A structural basis for variegating position effects. Cell 37:869–878
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Tong J, Lillico SG, Bi MJ, Qing T, Liu XF, Wang Y, Zheng M, Wang M, Dai YP, Whitelaw CB, Li N (2011) Tissue-specific and expression of porcine growth hormone gene in BAC transgenic mice. Transgenic Res 20:933–938
Uusi-Oukari M, Hyttinen J-M, Korhonen V-P, Vasti A, Alhonen L, Janne OA, Janne J (1997) Bovine aS1-casein gene sequences direct high-level expression of human granulocyte-macrophage colony-stimulating factor in the milk of transgenic mice. Transgenic Res 6:75–84
Wall RJ, Kerr DE, Bondioli KR (1997) Transgenic dairy cattle: genetic engineering on a large scale. J Dairy Sci 80:2213–2224
Zhimulev IF (1998) Polytene chromosomes, heterochromatin and position effect variegation. Adv Genet 37:1–566
Acknowledgments
The authors express gratitude to Dr. Nelly Khaidarova (Institute of Molecular Genetics, Academy of Sciences of Russia, Moscow, Russia) for help in preparation of recombinant DNA and Dr. Irina Gileva for providing us the recombinant human GM-CSF derived from E. coli. This study was financially supported by Grant-56.26 from Program of Fundamental Research RAN.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burkov, I.A., Serova, I.A., Battulin, N.R. et al. Expression of the human granulocyte–macrophage colony stimulating factor (hGM-CSF) gene under control of the 5′-regulatory sequence of the goat alpha-S1-casein gene with and without a MAR element in transgenic mice. Transgenic Res 22, 949–964 (2013). https://doi.org/10.1007/s11248-013-9697-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-013-9697-4