
Abstract
Xenogeneic skin, especially porcine skin, has already been used to cover large wounds in clinic practice of wound care. Our previous data showed that transgenic expression of human cytoxic T-lymphocyte associated antigen4-immunoglobulin (hCTLA4Ig) in murine skin graft remarkably prolonged its survival in xenogeneic burn wounds without extensive immunosuppression in recipients, suggesting that transgenic hCTLA4Ig expression in skin graft may be an effective and safe method to prolong its survival in xenogeneic wounds for coverage. Lentiviral transgenesis provides an extremely efficient and cost-effective method to produce transgenic animals. However, tissue-targeted transgenic expression of biologically functional protein by lentiviral transgenesis is rarely reported. In this work, a recombinant lentiviral vector (LV), named FKCW in this article, was constructed by inserting a skin-specific hCTLA4Ig expression cassette consisting of keratin 14 (K14) promoter, hCTLA4Ig coding sequence and an intronic fragment. Its efficacy for transgenesis and skin-specific expression of bio-active hCTLA4Ig protein was tested using mice as models. The LV FKCW was readily to be packaged and concentrated to high titres (1.287–6.254 × 109 TU/ml) by conventional lentivirus package system. Using eggs collected from only five mated females having been subjected to conventional super-ovulation treatment, 8 hCTLA4Ig transgenic founder mice were generated with the concentrated FKCW vector, and transgenic founder per injected and transferred egg was 6.3%, which was nearly 9-fold higher than that for DNA micro-injection with a similar transgene construct in our previous work. The lentiviral transgenic hCTLA4Ig exhibited strictly skin-specific expression at a level comparable to or even slightly higher than that of transgenic hCTLA4Ig delivered by micro-injection in a similar cassette. Lentiviral transgenic hCTLA4Ig protein remarkably suppressed human lymphocyte proliferation in vitro to a degree comparable to that of commercially purchased purified hCTLA4Ig protein with defined activity at similar concentrations. Besides, lentiviral hCTLA4Ig transgenic mouse skin grafted into rat burn wounds exhibited remarkably extended survival compared to wild-type skin of the same strain (13.8 ± 3.8 vs. 6.8 ± 3.0 days), indicating that lentiviral transgenic hCTLA4Ig did inhibit immune rejection against xenogeneic skin graft in vivo. These results laid down the foundation to further efficiently generate transgenic pigs skin-specifically expressing bio-active hCTLA4Ig by lentiviral transgenesis, and provided a demonstration that transgenic animals with tissue-targeted expression of biologically functional protein can be efficiently produced using LV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The exposed wounds are the primary threats to burn patients and can lead to infection and severe physiological complications (Orgill 2009). Early excision of necrotic burned tissues with skin grafting is often required for severe burn wounds (Orgill 2009; Desai et al. 1990; Wang et al. 2007). Autologous skin taken from unburned areas of patients is the ideal material for grafting, but the supply is limited in severely burned patients although expansion techniques such as “mesh” are available (Lari and Gang 2001; Vandeput et al. 1995). Allogeneic skin is the preferred substitute of autologous skin as a temporary coverage dressing (Orgill 2009), however, its disadvantages include ethic considerations, easier disease transmission and unstable supply due to the shortage of cadaveric donors (Weiner et al. 2010; Luo et al. 2005). Xenogeneic skin, especially porcine skin, has already been used to cover large wounds in clinic practice instead of allogeneic skin due to its similar physiological characteristics and plentiful supply (Weiner et al. 2010), but the severe immune rejection after transplantation limits its application (Luo et al. 2005).
Skin graft rejection is a typical cellular immune response mainly mediated by T cells. Cytoxic T-lymphocyte Associated Antigen4-Immunoglobulin (CTLA4Ig), which is a soluble chimeric molecule consisting of the extracellular domain of CTLA4 on the N-terminus and the constant and hinge regions of IgG on the C terminus, has proved to be effective in down-regulating T cell activation by blocking co-stimulatory pathway in different experimental models (Linsly et al. 1992; Jin and Xie 2003; Wang et al. 2004; Stell et al. 2003; Jin and Xie 2003 and Guo et al. 2003). Our previous study indicated that transgenic expression of CTLA4Ig in mouse skin prolonged its survival when grafted into rat burn wounds without extensive immuno-suppression in recipient rats (Wang et al. 2008), suggesting that transgenic CTLA4Ig expression in skin graft may be an effective and safe method to prolong xenogenic skin graft survival for the clinic practice of wound care.
Porcine skin is the most frequently used xenogeneic skin in clinic practice as a substitute for human allogeneic skin, therefore transgenic expression of hCTLA4Ig in porcine skin is of important clinic implication. Currently, the available methods for pig genetic modification include micro-injection, sperm-mediated gene transfer (SMGT), somatic cell nuclear transfer (SCNT) and lentiviral transgenesis. For transgenic pig production, micro-injection is highly inefficient, labor-intensive and cost-consumptive. SMGT is reported to be a simple, inexpensive and effective method for transgenic animal production, however, extremely variant data has been reported from different labs and the highly unstable outcome of this technology limits its application. SCNT has been successfully used to produce transgenic pigs in many cases, however, this method is still inefficient and labor-intensive, and plagued by high failure rate mainly due to perinatal death and unexpected health problems of survived cloned transgenic individuals. Besides, the antibiotic resistant gene, the necessary selection marker for transgenic nuclei donor cell culture which is finally added into the genomes of transgenic cloned individuals by SCNT process, brings additional uncertainties or complexities for clinic application of transgenic skin.
Lentiviral vector (LV), a retroviral vector which derives from lentivirus, is a well established gene-delivery vehicle primarily designed for gene therapy. The vectors of this kind are highly replication-defective by depleting of all viral structure genes only leaving the elements necessary for pseudo-type virus package. Besides, LV vectors are mutated to be “self-inactivating” by depleting a part of U3 area with no functional impact on vector (Myoshi et al. 1998). As a result, the promoter activity of long terminal repeat (LTR) is inactivated after retro-transcription and integration into host genome, and the transgene delivered by LV is only under transcriptional control of exogenous promoter inserted upstream of transgene, which further minimizes potential risks and makes transgene expression in a manner determined by the exogenous promoter. Taken together, LV is at least a theoretically safe vector suitable for transgenesis.
Data from different labs indicated that transgenesis with LV (lentiviral transgenesis) is an efficient and cost-effective approach to produce transgenic pigs or other mammalian animals. The transgene delivered by LV showed reliable, stable and inheritable transgene expression and no transcriptional silence through germline transmission. Besides, unlike prototypic retroviral vectors, LVs can transduce non-deviding cells and the provirus integration is independent on cell cycling, therefore transgene can be delivered directly into the genome of zygotes or pre-implantation early embryos by LV. LVs were first used to generate transgenic mice by transduction of murine 1-cell zygote or preimplantation embryos (Lois et al. 2002; Pfeifer et al. 2002) or embryonic stem cells (ESC) (Pfeifer et al. 2002). Compared to the traditional DNA microinjection technique, LV vector-mediated gene transfer resulted in a four- to eight- fold higher rate of transgenic animals per embryo treated (Pfeifer 2004), and more than 90% of F0-generation animals expressed the transgene (Lois et al. 2002; Pfeifer et al. 2002). Transgenic pig was the first transgenic farm animal generated with LVs. Using the LV containing eGFP to transduce porcine zygotes by perivitelline space injection (PSI), 31% of the injected/transferred porcine eggs resulted in transgenic founder pigs and 95% of founder pigs displayed green fluorescence (Whitelaw et al. 2004). Furthermore, with a LV containing eGFP under the drive of Keratin 14 (K14) promoter, lentiviral transgenic pigs displayed skin-specific eGFP expression (Hofmann et al. 2003), indicating that the LTR was completely inactivated and lentiviral transgene was only under the drive of exogenous promoter. Our recent data further showed that lentiviral transgene integrants with high expression in founders exhibited a relatively constant expression level per integrant over at least seven generations with no remarkable position effect observed (Wang et al. 2010). Besides, at least over seven generations the high-expressing integrants were not subjected to de novo methylation (Wang et al. 2010). These data suggested that transgenic lines with long-term stable transgene expression can be established by lentiviral transgenesis. Taken together, the current data suggested that lentiviral transgenesis may be a simple, reliable and highly efficient technique to establish a transgenic pig line exhibiting long-term stable and skin-specific overexpression of hCTLA4Ig as a reproducible source of xenogeneic skin grafts with extended survival for wound coverage.
Although lentiviral gene transfer is highly efficient, the majority of lentiviral transgenic animals currently produced were designed to express marker genes such as eGFP or siRNA molecules, and lentiviral tissue-specific expression of biologically functional proteins in vivo is rarely reported. In this study, based on a LV derived from HIV-1, we constructed a lentiviral skin-specific hCTLA4Ig expression vector by inclusion of an hCTLA4Ig expression cassette consisting of K14 promoter, intronic sequence and hCTLA4Ig coding sequence (CDS). The recombinant K14-CTLA4Ig LV, which is named FKCW in this article, was packaged into pseudo-typed viral particles with high tires (109–1010 TU/ml) by conventional “3rd generation retrovirus package system”. By infecting murine zygotes with the prepared lentiviral concentrate, transgenic mice displaying skin-specific hCTLA4Ig expression were efficiently generated, and the expression level was comparable to that of transgenic mice generated by DNA micro-injection with a similar hCTLA4Ig expression cassette in our previous study. Besides, lentiviral transgenic hCTLA4Ig protein exhibited full biological activity in vitro and prolonged skin graft survival in xenogeneic burn wounds. These results lay down the foundation to further generate transgenic pigs skin-speicifcally expressing bio-active hCTLA4Ig protein using lentiviral vector in an efficient and cost-effective manner, and provided a demonstration that transgenic animals with tissue-targeted expression of biologically active functional protein can be efficiently produced using LV.
Materials and methods
Lentiviral vector construct and preparation
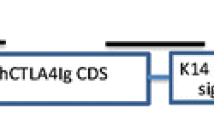
The LV for skin-specific CTLA4Ig expression was constructed using the vector FUGW containing an ubiquitine promoter-driven eGFP expression cassette as the backbone. The ubiquitine promoter in FUGW was replaced with a 2.0 kb K14 promoter fragment by PCR-based TA clone process, and the eGFP coding sequence (CDS) replaced with a 1.2 kb hCTLA4Ig cDNA fragment containing sequence coding hCTLA4Ig protein and a signal peptide. In addition, to stabilize transcripts and enhance transgene expression in vivo, an intronic sequence was inserted between K14 promoter and hCTLA4Ig CDS. The resulted recombinant plasmid was named FKCW.
FKCW plasmid was packaged intro pseudotyped lentiviral particles by conventional “three plasmid package system” (3rd Generation retrovirus package system) as previously described (Tiscornia et al. 2006). Briefly, 293FT cells were seeded in DMEM supplemented with 10% (v/v) fetal calf serum (FCS), 2 mM l-glutamine and 1 × MEM non-essential amino acids at a density of 2.5–3.8 ×104/cm2. The next day, cells were co-transfected with the plasmid psPAX2 (providing the viral structural proteins required for package), PMD2.0G (a VSV-G envelope protein expression plasmid) and the LV FKCW. After 7–11 h of culture, the media were replaced with fresh DMEM supplemented with 10% FCS. At 40 h post transfection, the culture supernatant was collected, centrifuged at 1,000 ×g for 5 min, filtered through a 0.45 μM filter unit, and further centrifuged at 25,000 ×g for 6 h at 4°C. The precipitate was resuspended with HBSS buffer, followed by sucrose gradient ultracentrifugation at 50,000 ×g for 90 min at 4°C. The final precipitate containing viral particles was resuspended in HBSS buffer and titred by Enzyme Linked Immunosorbent Assay (ELISA) using Quick Titre® Lentivral Quantitation Kit (Cell Biolabs, USA) as described in the manual.
Generation of transgenic mice
The outbred mouse strain KM was used as the egg donor. The mouse 1-cell eggs were collected and cultured as described (Hogan et al. 1994). The virus concentrate was thawed and briefly centrifuged to eliminate cell debris. 70–80 pl of virus suspension was injected into the peri-vitelline space of mouse eggs using a micro-injector guided by micro-manipulator (Narishega, Japan). The injected eggs were cultured in micro-drops of M16 media (Specialty Media, USA) covered with mineral oil (Sigma, USA) at 37°C, 5% CO2 in humidified atmosphere. The next day, cleaved 2-cell eggs were transferred into the oviducts of estrally synchronized recipient female mice. Transgenic founder mice were screened by PCR using four primer sets complementary to different regions of FKCW genome. The primers (P1 and P2) complementary to the lentiviral WPRE element were 5′-TGACTGGTATTCTTAACTATGTTGCTCC-3′ and 5′-AGGGAGATCCGACTCGTCTGA-3′, the primers to CTLA4Ig coding sequence (P3 and P4) were 5′-GACGCTGCTCAGTCTGGT-3′ and 5′-TGTAGAGTCCCGTGTCCAT-3′, the primers to the junction region between K14 promoter and intron (P5 and P6) were 5′-AGCCTGTGGGTGATGAAA-3′ and 5′-CAGACTGAGCAGCGTCCT-3′, and the primers to the flanking regions of intron were 5′-AGCCTGTGGGTGATGAAA -3′ and 5′-GGACCTCAGTGGCTTTGC-3′. The product sizes of the primer sets mentioned above were 537, 326, 389 and 525 bp, respectively. The transgenic individuals were bred in a closed colony to establish a transgenic inbred line.
Transgene expression analysis
Transgene expression was detected by RT-PCR, and western blot (WB). For RT-PCR, total RNA samples were prepared using Trizol reagent (Invitrogen,USA) and treated with DNaseI (Takara, Dalian China) to remove genomic DNA. Reverse transcription (RT) was performed with ReverTra Ace™ First Strand cDNA Synthesis Kit (TOYOBO, China) using Oligo (dT)20 as the primer as described in the manual. The sequences of primers used for PCR were 5′-GGACACGGGACTCTACATC-3′ and 5′-TACTCCTTGCCATTCAGC-3′ complementary to the extracellular region of human CTLA4 and the IgG region, respectively, and the PCR product was 432 bp. For WB, goat anti-human CTLA4 polyclonal antibody (Santa-cruz, USA) and HRP-conjugated goat anti-human IgG Fc antibody (Zongshan, China) were used as the primary and secondary antibodies, respectively. WB was performed as previously described (Wang et al. 2006).
Transgene expression was further quantitively analyzed by real-time PCR using the same primers for RT-PCR analysis. The real-time PCR system using SYBR PrimeScript RT-PCR Kit II(Takara,Dalian China) included SYBR Premix Ex Taq II 12.5 μl, PCR Primer(10 μM)1 μl each, template (reverse transcribed product of DNaseI-treated total RNA) 2 μl, and deionized water added up to 25 μl. PCR was performed with Bio-rad Real-time PCR system (Model CFX96), and the program was the following: 95°C 30 s; then entered cycles: 95°C 5 s, 60°C 30 s, plate read; run 39 more cycles, then melting curve from 55 to 95°C (reading every 0.5°C, holding 10 s). The liver cDNA samples were included as standard samples, and Rps18 as internal control.
Transgenic hCTLA4Ig protein bio-activity analysis
The immunosuppressive activity of lentiviral transgenic hCTLA4Ig protein was tested by one-way mixed lymphocyte culture (MLC) and skin grafting. For skin grafting, fifteen adult rats (regardless of gender) were randomly divided into three groups (five rats/each group). Each group was grafted with lentiviral transgenic, micro-injection transgenic or wild-type skin derived from the same mouse strain after wounds were made as previously described (Wang et al. 2008; Luo et al. 2005). For MLC, 1 ml of peripheral blood was harvested from lentiviral hCTLA4Ig transgenic mice from which serum samples containing hCTLA4Ig protein were prepared. The levels of CTLA4Ig protein in the serum samples were determined by ELISA as previously described (Wang et al. 2006). Human peripheral blood mononuclear cells (PBMCs) were isolated from two different individuals with no blood relations, which were marked A as reacting cells and B as stimulating cells, respectively. The stimulating cells (B) were inactivated by incubating with Mitomycin C at a final concentration of 25 μg/ml at 37°C, 5% CO2, humidified air for 0.5–1 h. Then the reacting cells (A) and the Mitomycin C treated stimulating cells (B) were mixed and cultured together in 96-well flat-bottomed plates in a total volume of 200 µl at 37°C, 5% CO2, humidified air for 5 d using RPMI 1,640 medium supplemented with 10% FBS, 50 IU/ml penicillin, 4 mM L-glutamin, 50 mg/ml streptomycin, 1 mM sodium pyruvate, and 0.05 mM β-mercaptoethanol. To test the bio-activity of transgenic hCTLA4Ig protein, different volumes of transgenic serum samples containing hCTLA4Ig protein collected from lentiviral hCTLA4Ig transgenic mice were added into the MLC system. Wild-type serum samples with no hCTLA4Ig protein collected from wild-type mice of the same strain were added into MLC system as negative controls, and purified hCTLA4Ig protein (R&D System, USA) dissolved into wild-type serum at different concentrations as positive controls. The MLC systems containing only reacting cells were used as a quality control for our MLC system. To test the level of reacting cell proliferation, 16 h before termination of culture the radioactive substrate 3H-TdR were added into MLC system at a dosage of 1uc per well, and the radioactivity of MLC was tested by radio-activity detector after culture was terminated.
Results
Efficient generation of transgenic mice skin-specifically expressing hCTLA4Ig using the LV FKCW
The constructed LV FKCW (showed in Fig. 1) was packaged into pseudotyped virus by conventional “three plasmid package system” and concentrated to high titres. Perivitelline space injection (PSI) of vector has emerged as the standard delivery method for lentiviral transgenesis in mammalian animals, but is limited by the need for high titre LV preparations. Therefore, the virus titre is the limiting factor for efficient lentiviral transgenesis in mammalian species. However, because the FKCW vector had no reporter gene, its actual biological titre can not be directly tested by fluorescence-activated cell sorting (FACS) using the infected cells. For accurate titration, FKCW was packaged and concentrated along with another LV FUGW which expressed eGFP from the same vector backbone. The physical titre of each vector was tested by ELISA assay in parallel. Then the biological titre of FUGW was tested by FACS using the infected 293FT cells as described (Tiscornia et al. 2006). Based on the physical titre of each vector and the biological titre of FUGW, the biological titre of FKCW was calculated using the following equation: FKCW biological titre (TU/ml) = (FKCW physical titre/FUGW physical titre) × FUGW biological titre (TU/ml). By this method, the actual biological titres of the FKCW virus concentrates were detected to be 1.287–6.254 × 109 TU/ml, which was sufficient for lentiviral transgenesis in mammalian animals by PSI.

Schematic representation of FKCW vector. This vector contains CMV promoter (CMVp), a central polypurine tract (cPPT), woodchuck hepatitis posttranscriptional regulatory element (WPRE) and self-inactivating 3′LTR. The CTLA4Ig expression is driven by K14 promoter, and an intronic sequence derived from rabbit β-globulin was inserted between K14 promoter and CTLA4Ig cDNA fragment for enhanced transgene expression in vivo. The arrows denoted as P1–P8 indicate the positions of primers used for screen of founder mice
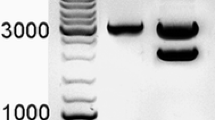
Transgenic mice were efficiently generated using the prepared FKCW virus concentrate. Five mated female mice subjected to conventional super-ovulation treatment using PMSG and HCG were used as egg donors from which 1-cell eggs were collected. For transduction of eggs, approximately 70–80 pl of the FKCW virus concentrate were injected into perivitelline space. Of all the 151 injected eggs, 127 cleaved into 2-cell, and the cleavage rate was 84.1% (127/151), which was comparable to that of untreated fresh eggs cultured in our lab. The injected 2-cell eggs were transferred into 4 recipient female mice, 3 recipient mice became pregnant and 13 pups were born. The development rate in vivo of the injected and transferred 2-cell eggs was 10.2% (13/127), which was nearly two-fold higher than that of the transferred 2-cell eggs subjected to DNA micro-injection with a similar hCTLA4Ig expression cassette in our previous work (Wang et al. 2005). Of the 13 pups, 8 were screened to be transgenic by PCR (Fig. 2). The rate of transgenic founder mice per injected and transferred egg was 6.3% (8/127), which was nearly 9-fold higher than that for DNA micro-injection with a similar transgene construct in our previous work (0.97%, 4/413) (Wang et al. 2005).

Analysis of integrity of the integrated genomes of FKCW vector in founder mice. The genomic DNAs of the thirteen founder mice were subjected to PCR using four pairs of primers corresponding to different regions of the genome of FKCW vector as showed in Fig. 1. The WPRE element (a), the CTLA4Ig coding sequence (b) and the junction region between K14 promoter and the intronic sequence (c) were all detected in the eight transgenic founder mice. To address whether the intronic fragment was spliced out from FKCW genomes during lentiviral transgenesis process, genomic DNAs of the founder mice were subjected to PCR using primers complementary to regions flanking the intron as showed in Fig. 1, and only one PCR product of more than 400 bp was detected in all the eight transgenic founder mice (d). M: DNA marker; 1–13: founder mouse genomic DNA samples
To test the integrity of the integrated provirus genomes of FKCW in founder mice, three pairs of PCR primers were designed to detect the presence of the WPRE element, CTLA4Ig CDS, and the junction region between K14 promoter and intronic sequence, respectively by PCR using genomic DNA of the founder mice as templates (Fig. 1). As shown in Fig. 2a–c, WPRE element, CTLA4Ig CDS and the junction region between K14 promoter and the intronic sequence were all detected in all the eight transgenic founder mice. To address whether the exogenous intronic fragment was spliced out from FKCW vector genomes during the process of lentiviral transgenesis, another pair of primers complementary to the regions flanking intron was designed (Fig. 1). If the intronic sequence was spliced out, the size of the PCR product would be 287 bp, or it would be 525 bp. As shown in Fig. 1d, only the PCR product of 525 bp was observed in the eight transgenic founder mice, suggesting that the exogenous intronic fragment was not spliced out during the lentiviral transgenesis process at least from the most of FKCW vector genomes in founder mice.
To detect the tissue distribution of lentiviral hCTLA4Ig expression, different organs (skin, liver, heart, lung, kidney, spleen and intestine) were subjected to RT-PCR, and Western blot (WB). The lentiviral transgenic hCTLA4Ig expression was detected to be skin-specific by the both two methods (Fig. 3a, b), which was consistent with our previous data for transgenic hCTLA4Ig delivered by micro-injection in a similar construct (Wang et al. 2006). Semi-quantitation of hCTLA4Ig expression was achieved by comparing the WB band densities of hCTLA4Ig to that of the internal control gene GAPDH, and data showed that the relative hCTLA4Ig expression level of the lentiviral transgenic mice was comparable to that of transgenic mice generated by micro-injection with a similar construct (0.63 vs. 0.57, Fig. 3c). For full-quantitation, real-time PCR was performed to detect transgenic hCTLA4Ig expression using house-keeping gene Rps 18 as the internal control. The Ct derivations and standard curves of the real-time PCR reactions for both transgenic hCTLA4Ig and Rps 18 indicated that the real-time PCR reactions were specific, and the log quantities of Ct values were linearly correlated with the respective template copies (Fig. 4a, b). Consistent with the data of WB assay, the expression level of lentiviral transgenic hCTLA4Ig detected by real-time PCR was comparable or even slightly higher than that of transgenic hCTLA4Ig delivered by micro-injection in a similar construct (Fig. 4c).

Tissue distribution and semi-quantitation of hCTLA4Ig expression. a The tissue-distribution of lentiviral transgenic hCTLA4Ig expression detected by RT-PCR. M: marker; 1–8: RNA samples prepared from lentiviral hCTLA4Ig transgenic mouse skin, heart, liver, spleen, lung, kidney, muscle and intestine; 9: RNA samples from skin of hCTLA4Ig transgenic mice previously produced by micro-injection with a similar transgene construct; b The tissue-distribution of lentiviral transgenic hCTLA4Ig expression detected by Western blot (WB). 1–7: protein samples prepared from heart, liver, spleen, lung, kidney, muscle and skin of lentiviral hCTLA4Ig transgenic mice; 8: protein sample from skin of hCTLA4Ig transgenic mice previously produced by micro-injection. c Semi-quantitation of hCTLA4Ig expression by comparing the WB band intensity of hCTLA4Ig to that of internal control gene GAPDH

Quantitative analysis of hCTLA4Ig expression in transgenic mouse skin by qRT-PCR. Total RNA samples were prepared from skins of hCTLA4Ig transgenic mice produced by lentiviral transgenesis and micro-injection, respectively. After DnaseI treatment, the total RNA samples were subjected to reverse transcription and real-time PCR analysis. a The melting and standard curves of the real-time RT-PCR reaction for hCTAL4Ig. b The melting and standard curves of the real-time PCR reaction for internal control Rps 18. c The relative expression levels of transgenic hCTLA4Ig in the skin of transgenic mice generated by lenviral transgenesis and mico-injection, respectively
Lentiviral transgenic hCTLA4Ig protein exhibited full biological activity
Because hCTLA4Ig was a soluble and secretory protein and would be transported into bloodstream after expression in skin, high levels of hCTLA4Ig protein were detected in the sera of lentiviral hCTLA4Ig transgenic mice, which was consistent with our previous data for hCTLA4Ig transgenic mice generated by micro-injection (Wang et al. 2006). The concentration of circulatory hCTLA4Ig protein in lentiviral transgenic mice were detected to be 11.87 µg/ml by Sandwich ELISA (Fig. 5).

Detection of circulatory hCTLA4Ig protein in lentiviral transgenic mouse sera by Sandwich ELISA. a The plate used to perform ELISA assay. Wild-type sera, lentiviral transgenic sera and the standard samples prepared by dissolving purified hCTLA4Ig protein with defined activity in wild-type sera at different concentrations (2, 4, 6, 8 and 10 μg/ml, respectively) were simultaneously subjected to ELISA assay in parallel. Each ELISA system had two duplicates, and the ELISA system with no sera was used as blank control. b Based on the data of standard samples (OD490 values of the wells containing standard samples), a standard curve was established of which the regression equation was: hCTLA4Ig concentration (μg/ml) = 28.23 × OD490 value −15.66. The mean OD490 values of the two wells containing lentiviral transgenic sera was 0.975, thereby the concentration of hCTLA4Ig protein in lentiviral transgenic mouse sera was estimated to be 11.87 μg/ml
To test the immunosuppressive activity of lentiviral transgenic hCTLA4Ig protein in vitro, different volumes of lentiviral transgenic mouse sera (10 and 20 µl, respectively) were added into the one-way mixed lymphocyte culture (MLC) system consisting of human PBMC collected from different individuals. Different concentrations (0.5, 1, 2 and 5 µg/ml) of commercially purchased purified hCTLA4Ig protein (R&D System, USA) with defined activity dissolved into wild-type mouse sera were included in MLC systems as positive controls, and the wild-type serum with no hCTLA4Ig protein as negative control. In addition, the reacting lymphocytes were cultured alone (without stimulating cells) as a quality control for our MLC system. As shown in Fig. 6, the reacting lymphocytes cultured alone exhibited the lowest proliferation, indicating that our MLC system was validated. The lentiviral transgenic mouse sera remarkably suppressed human lymphocyte proliferation in MLC system compared to the wild-type mouse serum without hCTLA4Ig protein (Fig. 6), indicating that lentiviral transgenic hCTLA4Ig protein was capable of inhibiting human lymphocyte activation. Besides, the two different final concentrations of lentiviral transgenic hCTLA4Ig protein in MLC system (0.59 µg/ml for 10 µl serum-added MLC and 1.18 µg/ml for 20 µl added) both suppressed human lymphocyte proliferation to a degree comparable to that of purified hCTLA4Ig protein at a similar final concentration in MLC (0.5 and 1.0 µg/ml, respectively) (Fig. 6), suggesting that the lentiviral transgenic hCTLA4Ig protein exhibited full biological activity in vitro.

Analysis of the biological activity of lentiviral transgenic hCTLA4Ig protein in vitro. One way mixed lymphocyte culture (MLC) consisting of human peripheral blood mononuclear cells (PBMCs) collected from different individuals was performed to test the biological activity of lentiviral transgenic hCTLA4Ig protein. 1–5: Purified recombinant hCTLA4Ig protein with defined activity was dissolved in wild-type sera and added into MLC system at different final concentrations (0.5, 1, 2 and 5 μg/ml, respectively) as positive controls, and wild-type sera with no hCTLA4Ig protein as negative control; 6 and 7: Different volumes of lentiviral transgenic sera (10 and 20 μl, respectively) were added into MLC system. 8: As a quality control for MLC system, the reacting cells were cultured alone without the presence of stimulating cells
To further test the biological activity of lentiviral transgenic hCTLA4Ig protein in vivo, skin grafting was performed from lentiviral hCTLA4Ig transgenic mice to rat burn wounds as previously described (Wang et al. 2008). Because human CTLA4 protein exhibits relatively higher cross-activity to that of rat (Lui et al. 2003), the impact of lentiviral transgenic hCTLA4Ig protein on rat response toward xenogenic skin graft should at least partly reflect its actual impact on human response. As shown in Fig. 7, severe rejection was observed in wild-type skin of the same mouse strain on 10 days post grafting, while in contrast the lentiviral hCTLA4Ig transgenic skin remained alive at this time point and no remarkable rejection was observed (Fig. 7a). The mean survival time (MST) of lentiviral hCTLA4Ig transgenic mouse skin grafted into rat burn wound was comparable to that of skin derived from hCTLA4Ig transgenic mouse generated by micro-injection (13.8 ± 3.8 vs. 12.9 ± 3.2 days), which was remarkably longer than that of wild-type skin (13.8 ± 3.8 vs. 6.8 ± 3.0 days) (Fig. 7b), indicating that lentiviral transgenic hCTLA4Ig protein did extended xenogeneic skin graft survival in vivo.

Lentiviral transgenic hCTLA4Ig protein prolonged skin graft survival in xenogeneic burn wounds. To test the bio-activity of lentiviral hCTLA4Ig protein in vivo, skin grafting was performed from lentiviral hCTLA4Ig transgenic mice to rats. a The skins grafted into rat burn wounds from lentiviral hCTLA4Ig transgenic mouse and wild-type mouse of the same mouse on 10 days post grafting, respectively. b The mean survival time (MST) of skins grafted from lentiviral hCTLA4Ig transgenic mice, hCTLA4Ig transgenic mice generated by micro-injection with similar construct and wild-type mice of the same strain
Discussion
Skin grafting is the primary treatment for care of large wounds. Currently, although different materials for wound coverage are available, considering the urgent requirements and the consequent complications, xenogeneic skin, especially porcine skin, is frequently used to cover large wounds as a substitute for human skin mainly due to similar physiological characteristics, plentiful supply and relatively easier control of disease transmissions compared to human skins collected from cadaveric donors (Weiner et al. 2010; Luo et al. 2005). However, the severe immune rejection after transplantation limits their application, and as a result the survival time of grafted xenogeneic skins is always too short to meet the clinical needs of burned patients without immunosuppression (Konigova et al. 2000). hCTLA4Ig is a well established molecule which is capable of down-regulating T cell activation and extending graft survival in different experimental models (Guerder and Flavell 1995; Lenschow et al. 1996). Our previous data showed that transgenic expression of hCTLA4Ig in skin graft effectively prolonged skin graft survival in xenogeneic burn wounds with no extensive immunosuppression in recipients (Wang et al. 2008), suggesting that transgenic expression of hCTLA4Ig in skin graft, instead of direct administration of hCTLA4Ig in recipients, may be an effective and safe method to extend xenogeneic skin graft survival for clinic purposes.
Considering the urgent requirements for wound coverage of patients with large burns and the consequent severe systemic immunosuppression after CTLA4Ig is extensively expressed in pigs (Phelps et al. 2009), establishment of a transgenic pig line with long-term stable and skin-specific expression of hCTLA4Ig may be the best choice for providing a reproducible resource of transgenic skin graft with extended survival for wound coverage. LV is an extremely efficient gene delivery vehicle primarily designed for gene therapy, and its efficacy for transgenesis has been well established in different mammalian species including pig. Our recent data further showed that a transgenic mammalian line with long-term stable transgene expression can be established by lentiviral transgenesis (Wang et al. 2010), suggesting that a transgenic pig line with long-term stable and skin-specific hCTLA4Ig expression can be established by this method in an efficient and cost-effective manner. To test this hypothesis, in this work a recombinant LV named FKCW which contains a skin-specific hCTLA4Ig expression cassette was constructed, and its efficacy for transgenesis was tested using mice as model animals.
The LV FKCW, which contains 3.4 kb inserted skin-specific hCTLA4Ig expression cassette, was readily to be packaged into high titre pseudotype virus by conventional lentivirus package process. The titre of concentrated FKCW virus suspension was between 1.287 and 6.254 × 109 TU/ml, which was sufficient for transgenic animal production by perivitelline space injection (PSI) with lentiviral concentrate. Since PSI has emerged as a standard method for lentiviral transgenesis in mammalian species due to its simplicity and easy operability (Pfeifer 2004), the readiness of FKCW to be packaged into high tire pesudotyped lenviral concentrate should make it an efficient vehicle to deliver the skin-specific hCTLA4Ig expression cassette into mammalian genomes. To test its efficacy for transgenesis, FKCW was used to produce transgenic mice by PSI. Results showed that using the eggs collected from only five conventionally super-ovulated female mice, eight hCTLA4Ig transgenic founder mice were produced by PSI with the FKCW lentiviral concentrate. Although the rate of transgenic founder per injected and transferred egg was 6.3%, the rate for pig may be higher because our unpublished data showed that, compared to murine eggs, much less porcine eggs collected from mated donor gilts and subjected to PSI with lentiviral concentrate were needed to be transferred into recipients for pregnancy. In this work, the number of treated and transferred murine eggs per recipient was 31.75. However, in our previous work, 12 porcine eggs collected, treated and transferred in the same way resulted into 6 piglets, of which 4 were transgenic, suggesting that less porcine eggs may be needed to produce hCTLA4Ig transgenic pigs using the FKCW vector, and higher efficiency may be obtained as a result.
The lentiviral hCTLA4Ig exhibited strictly skin-specific expression as detected by RT-PCR, and Western blot, indicating that the LTR of FKCW vector was completely inactivated after integration into host genome, and a transgenic pig line skin-specifically expressing hCTLA4Ig can be established using the FKCW vector. For enhanced transgene expression in vivo, exogenous intronic sequence is usually inserted between promoter and transgene CDS. However, lentiviral transgenesis is a complex biological process consisting of transcription, viral particle package, infecting and entering into target cells, reverse transcription and provirus integration into host genome. Thus, one question remains to be answered that whether the exogenous intronic sequence inserted into LV would be spliced out from LV genome during the process of lentiviral transgenesis. In this work, for enhanced hCTLA4Ig expression in vivo, the skin-specific hCTLA4Ig expression cassette inserted into FKCW vector contained an intronic sequence derived from rabbit β-globin gene. Data showed that the FKCW genome without the exogenous intronic sequence was not detected in any of the eight transgenic founders, indicating that the exogenous intron was not spliced out from at least the most of FKCW vector genomes during lentiviral transgenesis process. Besides, the lentiviral transgenic hCTLA4Ig exhibited a expression level comparable to that of transgenic hCTLA4Ig in a expression cassette containing the same promoter and intronic fragment delivered into the same mouse strain by micro-injection in our previous work, further indicating that the exogenous intron inserted into FKCW vector was not spliced out.
The lentiviral transgenic hCTLA4Ig exhibited biological activity as tested by both one-way MLC assay using human PBMCs and skin grafting using rats as recipients. In MLC assay, the lentiviral transgenic hCTLA4Ig protein suppressed human lymphocyte proliferation to extents comparable to those of the commercially purchased purified recombinant hCTLA4Ig protein with defined activity at similar final concentrations in MLC system. In skin grafting, lentiviral hCTLA4Ig transgenic skin displayed remarkably extended survival compared to wild-type skin derived from the same mouse strain, and comparable survival compared to hCTLA4Ig transgenic skin derived from transgenic mice produced by micro-injection with similar expression cassette. Because human CTLA4 exhibits relatively higher cross-reactivity to that of rat (Lui et al. 2003), the impact of lentiviral transgenic hCTLA4Ig on xenogeneic skin graft survival in rat wounds should at least partly reflect its actual impact on that for human. These results further indicated that FKCW vector can be used to generate transgenic pigs skin-specifically expressing bio-active hCTLA4Ig protein.
This work provides a demonstration that transgenic animals with tissue-targeted expression of biologically active functional protein can be efficiently produced by lentiviral transgenesis. For this work, generation of a transgenic pig line skin-spcifically expressing bio-active hCTLA4Ig protein as a reproducible resource of xenogeneic skin with extended survival for wound coverage is the ultimate goal. Lentiviral transgene was primarily designed to be used for gene therapy which was aimed to be administered directly into patients. To improve the safety of lentiviral transgene, LV is designed to be highly replication-defective by depleting of all viral structural or functional genes and only leaving the necessary elements for pseudotype virus package. Besides, the LTR of LV is mutated to be self-inactivated after provirus integrated into host genome by depleting a part of U3 area (Myoshi et al. 1998), which not only minimizes the risks related to endogenous gene activation, but also makes the expression of lentiviral transgene only under the control of exogenous promoter inserted upstream the transgene. Therefore, the main concerns for the safety of lentiviral transgene are the risks related to the random integration process of provirus, and not the LV itself. For the ultimate purpose of this work, lentiviral transgenic hCTLA4Ig is not aimed to be administered directly into patients, but delivered into skin graft instead, and then the lentiviral transgenic skin is aimed to be grafted into the wounds of patients for coverage. Therefore, the provirus integration process does not occur in patients, but in skin donor instead. Considering the high replication-defectiveness of LV and the skin-specific expression pattern of hCTLA4Ig, lentiviral hCTLA4Ig transgenic skin should be at least theoretically safe for clinic application. However, reasonably designed experiments are needed to test the actual safety of lentiviral transgenic skin.
References
Desai MH, Herndon DN, Broemeling L et al (1990) Early burn wound excision significantly reduces blood loss. Ann Surg 211:753–759
Guerder S, Flavell RA (1995) Co-stimulation in immune tolerance and autoimmunity. Int Rev Immunol 1995(13):135–146
Guo L, Fujino M, Kimura H, Funeshima N, Kitazawa Y, Harihara Y, Tezuka K, Makuuchi M, Suzuki S, Li XK (2003) Simultaneous blockade of co-stimulatory signals, CD28 and ICOS, induced a stable tolerance in rat heart transplantation. Transpl Immunol 12(1):41–48
Hofmann A, Kessler B, Ewerling S et al (2003) Efficient transgenesis in farm animals by lentiviral vectors. EMBO Rep 4:1054–1060
Hogan B, Beddington R, Costantini F, Lacy B (1994) Manipulating the mouse embryo: a laboratory manual, second edition. Cold Spring Harbor Laboratory Press
Jin YZ, Xie SS (2003) Bicistronic adenovirus-mediated gene transfer of CTLA4Ig gene and CD40Ig gene result in indefinite survival of islet xenograft. Transpl Proc 35:3165–3166
Konigova R, Matouskova E, Broz L (2000) Burn wound coverage and burn wound closure. Acta Chir Plast 42:64–68
Lari AR, Gang RK (2001) Expansion technique for skin grafts (Meek technique) in the treatment of severely burned patients. Burns 27:61–66
Lenschow DJ, Walunas TL, Bluestone JA (1996) CD28/B7 system of T cell co-stimulation. Annu Rev Immunol 14:233–258
Linsly PS, Wallence PM, Iohnson J (1992) Immuno suppression in vivo by a soluble form of the CTLA4 T cell activation molecule. Science 257:792–797
Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295:868–872
Lui VCH, Tam PKH, Leung MYK et al (2003) Mammary gland-specific secretion of biologically active immunosuppressive agent cytoxic-T-lymphocyte antigen 4 human immunoglobulin fusion protein (CTLA4Ig) in milk by transgenesis. J Immunol Method 277:171–183
Luo GX, Wu J, Chen XW et al (2005) CTLA4Ig introduced by adenoviral vector locally to prolong the survival of xenogeneic skin grafts on rat burn wounds. J Trauma 59:1209–1215
Myoshi H, Blomer U, Takahashi M, Gage FH, Verma IM (1998) Development of a self-inactivating lentivirus vector. J Virol 72:8150–8157
Orgill DP (2009) Excision and skin grafting of thermal burns. New Engl J Med 360:893–901
Pfeifer A (2004) Lentiviral transgenesis. Transgenic Res 13:513–522
Pfeifer A, Ikawa M, Dayn Y, Verma IM (2002) Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc Natl Acad Sci USA 99:2140–2145
Phelps CJ, Ball SF, Vaught TD, Vance AM, Mendicino M, Monahan JA, Walters AH, Wells KD, Dandro AS, Ramsoondar JJ, Cooper DK, Ayares DL (2009) Production and characterization of transgenic pigs expressing porcine CTLA4Ig. Xenotransplantation 16:477–485
Stell D, Marshall H, Bradley JA, Bolton EM (2003) CTLA4-Ig abrogates the anti-globulin response and prolongs cardiac allograft survival after anti-CD2 treatment. Transpl Immunol 12(1):1–7
Tiscornia G, Singer O, Verma IM (2006) Production and purification of lentiviral vectors. Nature Protocls 1:241–245
Vandeput J, Nelissen M, Tanner JC, Boswick J (1995) A review of skin meshers. Burns 21:364–370
Wang YF, Xu AG, Hua YB, Wu WX (2004) Effect of local CTLA4Ig gene transfection on acute rejection of small bowel allografts in rats. World J Gastroenterol 10(6):885–888
Wang Y, Wang FC, Wei H, Ni Y, Wu J, Gao X (2005) Establishement of transgenic mouse line skin-specifically expressing hCTLA4Ig. J Genet Genomes 32:916–922
Wang Y, Ni Y, Wei H, Wang FC, Ge L-P, Gao X (2006) Stable skin-specific overexpression of human CTLA4Ig in transgenic mice through seven generations. Acta Biochim Biophys Sin 38:171–178
Wang YB, Ogawa Y, Kakudo N, Kusumoto K (2007) Survival and wound contraction of full-thickness skin grafts are associated with the degree of tissue edema of the graft bed in immediate excision and early wound excision and grafting in a rabbit model. J Burn Care Res 28:182–186
Wang Y, Wei H, Yong N, Ge LP, Liu Q, Mao XL, Zhao YJ, Wu J (2008) Transgenic expression of cytoxic T lymphocyte associated antigen 4-immunoglobulin prolongs xenogeneic skin graft survival without extensive immunosuppression in rat burn wounds. J Trauma 65:154–162
Wang Y, Song YT, Liu Q, Liu CE, Wang LL, Liu Y, Zhou XY, Wu J, Wei H (2010) Quantitative analysis of lentiviral transgene expression in mice over seven generations. Transgenic Res 19:775–784
Weiner J, Yamada K, Ishikawa Y et al (2010) Prolonged survivial of GalT-KO swine skin on baboons. Xenotransplantation 17:147–152
Whitelaw CBA, Radcliffe PA, Ritchie WA et al (2004) Efficient generation of transgenic pigs using equine anaemia virus (EIAV) derived vector. FEBS Lett 571:233–236
Acknowledgments
This work was supported by grants from National Basic Research Program of China (2007CB513007), National Natural Science Foundation of China (30700076) and Chongqing Natural Science Foundation (CSTC, 2009BB5318). We’d like to thank Dr. Baltimore of California Institute of Technology (USA) for his kind offer of the lentiviral vector FUGW used in this study.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jiang, W., Zhou, Xy., Wang, Ll. et al. Skin-specifically transgenic expression of biologically active human cytoxic T-lymphocyte associated Antigen4-Immunoglobulin (hCTLA4Ig) in mice using lentiviral vector. Transgenic Res 21, 579–591 (2012). https://doi.org/10.1007/s11248-011-9559-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-011-9559-x