
Abstract
Certain types of human papillomaviruses (HPV) are causatively associated with cervical carcinoma, the second most common cancer in women worldwide. Due to limitations in the availability of currently used virus-like particle (VLP)-based vaccines against HPV to women of developing countries, where most cases of cervical cancer occur, the development of a cost-effective second-generation vaccine is a necessity. Capsomeres have recently been demonstrated to be highly immunogenic and to have a number of advantages as a potential cost-effective alternative to VLP-based HPV vaccines. We have expressed a mutated HPV-16 L1 (L1_2xCysM) gene that retained the ability to assemble L1 protein to capsomeres in tobacco chloroplasts. The recombinant protein yielded up to 1.5% of total soluble protein. The assembly of capsomeres was examined and verified by cesium chloride density gradient centrifugation and sucrose sedimentation analysis. An antigen capture enzyme-linked immunosorbent assay confirmed the formation of capsomeres by using a conformation-specific monoclonal antibody which recognized the conformational epitopes. Transplastomic tobacco plants exhibited normal growth and morphology, but all such lines showed male sterility in the T0, T1 and T2 generations. Taken together, these results indicate the possibility of producing a low-cost capsomere-based vaccine by plastids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cervical cancer is the second most common cancer in women worldwide, with approximately 493,000 new cases and 274,000 deaths occurring annually around the globe (Parkin and Bray 2006). Human papillomavirus (HPV) types 16 and 18 have been identified to be responsible for approximately 70% of invasive cervical cancers, with HPV-16 being by far the most prevalent type and found in about 54% of all cervical cancer cases (Smith et al. 2007).
L1 is the major capsid protein of HPV and self assembles into higher order molecular structures, such as capsomeres and virus-like particles (VLPs). Each VLP consists of 72 capsomeres, and each capsomere is a pentamer of the L1 protein arranged in an icosahedral surface lattice (Baker et al. 1991). The formation of disulphide bonds between L1 protein molecules in adjacent capsomeres is essential for VLP stability, and the reduction of these bonds leads to the disassembly of VLPs into capsomeres (Sapp et al. 1995). Replacement of either of the two highly conserved cysteines in L1 protein by serine prevents VLP assembly and leads to the retention of L1 capsomeres (Sapp et al. 1998).
VLPs are highly immunogenic as they can induce high titres of neutralizing antibodies (Rose et al. 1998) that have been shown to successfully prevent HPV infections (Koutsky et al. 2002; Villa et al. 2006). The first two VLP-based vaccines using L1 protein as the selected antigen have recently been introduced onto the market (Harper et al. 2004; Villa et al. 2006; Müller and Gissmann 2007). One of these vaccines, obtained from a yeast expression system, is a tetravalent that covers HPV types 16, 18, 6 and 11, while the second is a bivalent vaccine covering HPV types 16 and 18 and is obtained from insect cells. However, relatively high production and distribution costs are associated with these two vaccines. Furthermore, both require a continuous cold chain and sterile needles for intramuscular administration. Consequently these vaccines are cost-intensive and, as such, they will likely be unavailable (=unaffordable) for people in developing countries where more than 80% of all cervical cancer cases occur (Parkin and Bray 2006; Stanley et al. 2008). Therefore, there is an urgent need for second-generation cost-effective vaccines (Stanley et al. 2008).
Capsomeres represent a potentially cost-saving alternative to VLPs, as they are considered to be thermo-stable, which is advantageous for use in developing countries where cold chains are difficult to maintain (Stanley et al. 2008). Capsomeres have been shown to induce high titres of neutralizing antibodies and L1-specific cytotoxic T-lymphocytes (CTLs) upon oral, intranasal and subcutaneous immunization, and they have also been reported to protect dogs against viral challenge (Rose et al. 1998; Fligge et al. 2001; Yuan et al. 2001; Öhlschläger et al. 2003; Dell et al. 2006; Thönes and Müller 2007; Schädlich et al. 2009a).
The use of plants as an alternate system for the production of biopharmaceuticals, therapeutic proteins and recombinant vaccines is associated with a number of advantages, such as low cost, ease to scale up for mass production and low health risks; there is also the additional potential benefit that the products thus produced can be administered unprocessed (e.g plant leaves) or partially processed material (Daniell et al. 2001; Fischer et al. 2004; Ma et al. 2005). HPV-16 L1 protein has been expressed in plants following integration of the transgene into the nuclear genome (Biemelt et al. 2003; Varsani et al. 2003; Liu et al. 2005; Maclean et al. 2007). However, in all of these studies, the antigen expression level was <1% of total soluble protein (TSP) with the exception of that of Maclean et al. (2007), who obtained 11% TSP by expressing a human codon-optimized gene linked to a chloroplast-targeting signal. The low level of antigen expression (an average of 0.01–0.4% of TSP; Daniell et al. 2001) is one of the main drawbacks of transgene integration into the nuclear genome.
This constraint can be solved by engineering the plastid genome, which in terms of developing applications involving the production of plant-made pharmaceuticals has many advantages, including high levels of transgene expression due to high copy number, absence of epigenetic effects, transgene containment via maternal inheritance and multi-gene expression in a single transformation event (for review, see Maliga 2002; Bock 2007; Koop et al. 2007; Chebolu and Daniell 2009). Various vaccine antigens against several human and animal diseases have been successfully expressed in plastids, with an average expression level in the range of 4–31% of TSP (Chebolu and Daniell 2009). The highest transgene expression in chloroplasts obtained to date is 70% of TSP for an antibacterial lysin (Oey et al. 2009). In two recent studies, HPV-16 L1 VLPs were expressed in tobacco chloroplasts, obtaining 24% (Fernandez-San Millan et al. 2008) and 1.5% (Lenzi et al. 2008) of TSP, respectively. These VLPs obtained from the chloroplast were subsequently shown to be immunogenic in mice (Fernandez-San Millan et al. 2008).
The aim of the study reported here was to demonstrate the successful production of capsomeres in plastids through the expression of a mutated L1 gene (L1_2xCysM) having two cysteines replaced by serines. We found that the expression of the L1_2xCysM gene confined the assembly of L1 protein to pentameric capsomeres. The assembly and correct formation of capsomeres was confirmed by sucrose gradient analysis and further verified by an enzyme-linked immunosorbent assay (ELISA), using a conformation-specific antibody. The considerable expression of the modified L1 protein and its assembly into capsomeres makes this plastid expression system suitable for developing a cost-effective second-generation vaccine against HPV.
Materials and methods
Vector construction
A precursor vector pPNG1014_MCS120 was constructed as introduced by Ye et al. (2001), with slight modifications. This plasmid contained the plastid-encoded polymerase (PEP) promoter from the rrn 16 gene (Prrn; Svab and Maliga 1993) and the ribosomal binding site (RBS) from the leader sequence of gene 10 (G10L) of the lambda phage T7 (Studier et al. 1990). Nuclear encoded polymerase (NEP) promoter (Prrn −62NEP; Hajdukiewicz et al. 1997) was fused with PrrnPEP immediately downstream, followed by G10L fusion. Thereafter, the sequence encoding for first 14 amino acids of the green fluorescent protein (GFP) (GFP14) was fused with the 5′ end of the gene of interest. Cleavage space was left for the multiple cloning site (MCS), and the 5′-untranslated region (5′ UTR) was linked to the gene of interest to allow downstream insertion of the aadA gene. This 5′ UTR consisted of synthetic ribosomal binding site.
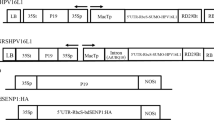
The mutated L1 gene (L1_2xCysM), in which two cysteines, 175 and 428, were replaced by serine residues (Schädlich et al. 2009b), was amplified by PCR using forward primer (oli) 5′-AAAAgctagcATGTCTACTTGCCTCCTGTC-3′ and reverse primer 5′-TTTTTTgcatgcTTACAGCTTACGTTTTTTGC-3′ (restriction sites in lower case). The L1_2xCysM gene was cloned in pPNG1014_MCS120 by ClaI and MluI to obtain plasmid pPNGL1M. Plasmid pT7PHB-N (Lössl et al. 2005) was used to obtain the final transformation vector pPNGL1MT. Plasmid pT7PHB-N contained the aadA gene (conferring resistance to spectinomycin and streptomycin), the terminator from the large subunit of ribulose-bisphosphate carboxylase gene (TrbcL) from Chlamydomonas reinhardtii and the flanking regions INSL and INSR, homologous to the loci trnN and trnR, respectively, in the inverted repeats (IR) of the tobacco plastome. The flanking sequences included nucleotides 109,230–110,348 and 110,349–111,520 of IR-A as well as nucleotides 131,106–132,277 and 132,278–133,396 of IR-B. Plasmid pT7PHB-N was cut with SacII and BamHI, and the whole cassette from PNGL1M was cloned in pT7PHB-N by SacII and BglII. The figure in the Electronic Supplementary Material (ESM) shows the vector construction pattern. Nucleotide positions for transgene insertions are given according to the plastome sequence data for EMBL accession no. Z00044, as initially reported by Shinozaki et al. (1986) and later updated by Wakasugi et al. (1998) and Yukawa et al. (2005). All cloning procedures were carried out using the standard methods described by Sambrook et al. (1989).
Transformation and regeneration of transplastomic plants
Nicotiana tabacum (Nt) cv. Petit Havana plantlets (Surrow Seeds, Sakskøbing, Denmark) were grown from seeds in vitro at 25°C (light intensity: 0.5–1 W/m2 Osram L85 W/25 universal-white fluorescent lamps) on agar-solidified MS medium (Murashige and Skoog 1962) containing MS salts and sucrose (30 g/l). Leaves were transformed using a particle gun (PDS1000He; Bio-Rad, Hercules, CA) by coating plasmid DNA on gold particles (0.6 μm; Svab et al. 1990). Following bombardment, leaves were cut into 5-mm pieces and placed on RMOP medium containing 500 mg/l spectinomycin (Svab et al. 1990). After four to six cycles of shoot regeneration on the same medium with spectinomycin, shoots were transferred to B5 medium (Gamborg et al. 1968) containing spectinomycin, for rooting. Transformed plants were then shifted to the greenhouse for further studies and seed production. These plants were pollinated with pollen from wild-type plants and seeds were collected. Seeds from the T0 generation were grown on agar-solidified MS medium (with MS salts and 30 g/l sucrose) containing 500 mg/l spectinomycin to generate T1 progeny. The T2 generation was obtained in the same way from the seeds of T1 progeny.
Confirmation of transgene integration
The PCR assay was performed to confirm transgene integration within the plastome in the T0 and T1 generations. DNA was extracted from 100–150 mg of plant leaves by the cetyltrimethylammonium bromide (CTAB) procedure (Murray and Thompson 1980). To confirm correct insertion of the L1_2xCysM gene within the plastid genome, PCR was carried out with the forward primer oli252 (positioned within the plastome outside the vector flanks; sequence 5′-AGACAGCGACGGGTTCTCTG-3′) and reverse primer oli248 (within L1_2xCysM gene; sequence 5′-GTACTTGGGGATCCTTTGCC-3′). For confirmation of the correct insertion of the aadA gene within the plastid genome, forward primer oli251 (5′-CCAGTATCAGCCCGTCATAC-3′) located within the aadA gene and reverse primer oli253 (5′-GATCCGAGCCATAGAATTTC-3′) located in the plastid genome outside the vector flanks were used. All primer positions are shown in the ESM (figure).
Western blot analysis
Soluble proteins were extracted from leaf discs collected from the plants grown under sterile conditions. Leaf samples (100 mg) were ground in liquid nitrogen, homogenized in sodium dodecyl sulphate (SDS) buffer (187.5 mM Tris-HCl, pH 6.8, 6% SDS, 30% glycerol, 15% ß-mercaptoethanol, 0.03% bromophenol blue), and the extract heated at 95°C for 5 min. The proteins were separated by SDS–polyacrylamide gel electrophoresis (12.5% polyacrylamide gel), transferred onto nitrocellulose membrane (Hybond C; GE Healthcare, Little Chalfont, UK) and blocked with 5% skimmed milk for 30 min. The membrane was incubated for 1 h with HPV-16 L1- specific monoclonal antibody MD2H11 (DKFZ, Heidelberg, Germany) diluted 1:4,000 in 5% skimmed milk, washed three times (10 min each time) with PBS containing 0.3% of Tween-20 (PBS-T) and incubated a second time for 1 h with peroxidase-conjugated goat antimouse immunoglobulin G (IgG; Sigma, St. Louis, MO) as secondary antibody diluted 1:3,000 in skimmed milk. After washing the membrane with PBS-T, proteins were detected by chemiluminescence, and the bands were visualized on X-Ray film (Kodak). Fractions from a sucrose gradient and CsCl centrifugation were also analyzed as described above.
Antigen capture ELISA
Tobacco leaves were obtained from the transgenic plants grown in sterile conditions. Leaves (100 mg) were ground in liquid nitrogen and homogenized in extraction buffer containing 5 mM MgCl2, 5 mM CaCl2, 1 M NaCl, 0.01% Triton X-100, 20 mM Hepes, pH 7.4, and 1 mM phenylmethylsulfonyl fluoride (PMSF). The extracts were centrifuged at 13,000 rpm for 5 min and the supernatant collected. Each well of 96-well microtitre plates was coated overnight at 4°C with 50 μl of HPV-16 L1 conformation specific mouse monoclonal antibody Ritti01 (Thönes et al. 2008) diluted 1:300 in PBS. The plates were washed with PBS-T and blocked for 1 h at 37°C with a blocking solution containing PBS, 3% skimmed milk and 0.3% Tween-20 (PBS-TM). Plant extracts were added to the wells and incubated for 1 h at 37°C. After three washing steps, 50 μl of polyclonal rabbit antiserum (1:3,000 in PBS-TM) raised against HPV-16 L1 was added to each well, and the plates were incubated for 1 h at 37°C. The plates were then washed three times, incubated for 1 h at 37°C after the addition of 50 μl of goat anti-rabbit peroxidase conjugate (Sigma; 1:3000 in PBS-TM) to each well and were washed thoroughly before 100 μl of staining solution {100 mM sodium acetate, 44 mM NaH2PO4, pH 4.2, 1 mg ABTS [2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)]/ml, 0.012% H2O2} was added to each well. After 10–20 min, the measurement was carried out at 405 nm. Fractions from a sucrose gradient and CsCl centrifugation were analyzed in the same manner as described above.
Sucrose gradient sedimentation analysis of protein
Tobacco leaves (500 mg) were ground in liquid nitrogen and homogenized with extraction buffer (5 mM MgCl2, 5 mM CaCl2, 1 M NaCl, 0.01% Triton X-100, 20 mM Hepes, pH 7.4, 1 mM PMSF). The proteins were extracted by sonication for 30 s. The lysate was cleared by centrifugation at 13,000 rpm for 15 min at 4°C, loaded onto a linear 5–50% (w/v) sucrose gradient in extraction buffer and centrifuged at 36,000 rpm for 3 h at 4°C using a Beckman SW41 Ti rotor (Beckman Coulter, Brea, CA). A total of 20 fractions (600 μl each) were collected from the bottom of the tube, and their refractive indices were determined. These fractions were then analyzed by Western blotting and antigen capture ELISA.
Protein purification by cesium chloride gradient centrifugation
Leaf material (5 g) from transgenic plants was ground in liquid nitrogen and homogenized with extraction buffer (5 mM MgCl2, 5 mM CaCl2, 1 M NaCl, 0.01% Triton X-100, 20 mM Hepes, pH 7.4, and 1 mM PMSF). After sonication, the lysate was cleared by centrifugation at 17,000 rpm for 30 min at 4°C. The cleared lysate was transferred onto a two-step gradient consisting of 7 ml of sucrose (30% w/v) on top of 7 ml of CsCl (58% w/v), and then centrifuged at 27,000 rpm for 3 h at 10°C. The interphase between sucrose and the CsCl was mixed with a CsCl layer and centrifuged at 50,000 rpm for 16 h at 20°C using a Sorvall TFT 65.13 rotor (Thermo Fisher Scientific, Waltham, MA). A total of 14 fractions (1 ml each) were collected from the bottom of the tube and analyzed by ELISA. A small volume of fractions was dialyzed against distilled water using a filter with a pore size of 0.025 μm (Millipore, Bedford, MA) and used for Western blot analysis.
Results
Chloroplast transformation vectors
The constructed plastid transformation vector PNGL1MT contained the mutated L1 gene (L1_2xCysM). This gene was expressed by a cassette containing the promoters for the plastid and nuclear-encoded polymerases (PrrnPEP and Prrn −62NEP, respectively) in combination with the terminator sequence from the large subunit of the ribulose-bisphosphate carboxylase gene (TrbcL) from C. reinhardtii. The DNA sequence for the first 14 amino acids of the GFP protein (GFP14) were fused with the transgene to enhance expression, as demonstrated by Ye et al. (2001). The aadA gene was used to select transplastomic plants. The PrrnPEP-Prrn −62NEP promoter served as a bicistronic promoter, driving the expression of both the L1_2xCysM and aadA genes. The trnN and trnR loci were used for homologous recombination within the tobacco plastid genome. The figure in the ESM shows the complete construct within the tobacco chloroplast genome, including the primers used to confirm the insertion of the transgenes within the plastid genome.
Plastid transformation and morphology of transplastomic plants
Tobacco leaves were transformed by the biolistic method and then placed on RMOP medium containing 500 mg/l spectinomycin for selection and regeneration. Control plants and all untransformed explants bleached out, while green microcalli developed from transformed explants that continued to grow on selection medium. Homoplastomy was achieved by subjecting resistant shoots to three regeneration cycles under the same conditions. For rooting, transgenic shoots were placed on B5 selection medium and then transferred to the greenhouse into pots.
All transplastomic lines transformed with the L1_2xCysM gene were male sterile and shed flowers (Fig. 1). Flowers were produced but showed senescence before maturity and did not produce seeds. However, plants had normal growth and morphology, like those of the wild type. Male sterility and flower senescence persisted in the successive T1 and T2 generations obtained by pollinating transgenic plants with wild-type plants.

A branch of transplastomic tobacco plant showing senescence of flowers and male sterility. Arrows on the lower section of branch Marks of shed flowers, upper part of figure flowers without seeds
Confirmation of transgene integration in transgenic plants
Tobacco plants with the L1_2xCysM gene were analyzed for transgene integration in the plastid genome by PCR. The primers oli248 (within L1_2xCysM) and oli252 (within cp at 5′ end) amplified a 1,939-bp DNA fragment, as expected for L1_2xCysM. This PCR-generated fragment confirmed the correct insertion of L1_2xCysM at the right insertion site (INSR) within the plastome. To prove the correct insertion of the aadA gene at the INSL within the plastome, a PCR was carried out with primers oli251 (within aadA) and oli253 (within chloroplast at 3′ end), amplifying the expected 1,981-bp fragment. Figure 2 depicts the amplified products from both flanks. Similar results were obtained by the PCR analysis of T1 plants (data not shown). Seeds of the T1 and T2 generations uniformly germinated on spectinomycin (500 mg/l) containing MS medium, as expected for homoplastomic plants.

PCR analysis of the transgenes inserted within the plastome. a Amplification of L1_2xCysM gene (1,939 bp) with the primers oli248 and oli252. b Amplification of the aadA gene (1,981 bp) with the primers oli251 and oli253. Three independently generated transplastomic lines (Lanes 1, 2, 3) were analyzed. M Marker, WT wild type
Expression and quantification of the L1 protein
Expression of recombinant L1 protein was detected by Western blot using MD2H11 antibody specific for HPV-16 L1 protein (Schädlich et al. 2009a). Three independently generated transplastomic plant lines were analyzed (Fig. 3a, lanes 1–3). Each line showed an expected band of 56.5 kDa corresponding to the calculated size of GFP14-L1 protein (Fig. 3a). Baculovirus-derived purified VLPs (DKFZ) were used as the positive control. No protein was detected in samples extracted from wild-type tobacco control plants. All three transplastomic lines generated signals in the antigen capture ELISA using the conformation-specific monoclonal antibody Ritti01 (Fig. 3b), thereby demonstrating the presence of conformational epitopes in chloroplast-derived L1 protein. VLPs and samples from wild type served as positive and negative controls, respectively. In addition, PBS was used as a negative control for each sample in ELISA.

L1 protein expression and quantification. a Western blot indicating the expression of plant-derived L1 protein (56.5 kDa) from three independent transplastomic lines in comparison with baculovirus-derived purified virus-like particle (VLPs). MD2H11 monoclonal antibody specific for HPV-16 L1 protein was used. Wild-type (WT) protein served as the control. Lanes 1, 2, 3 Protein samples from three independent transplastomic lines (transgenic lines 1, 2, 3, respectively). b Antigen capture enzyme-linked immunosorbent assay (ELISA) of the three transplastomic lines showing the accumulation of L1 protein in leaf extracts. VLPs were used as the positive control. Monoclonal antibody Ritti01 was used for the detection of conformational epitopes. Protein extracted from WT leaves and phosphate buffered saline (PBS) (for each sample) were the negative controls. c Protein quantification by western blot analysis based on a dilution series of baculovirus-derived L1 VLPs as reference (2, 4 and 8 ng). Total soluble leaf protein from transplastomic line 1 (1, 2 and 3 μg) was loaded in lanes. Monoclonal antibody MD2H11 was used
The amount of L1 protein was quantified by Western blotting using the MD2H11 antibody (Fig. 3c) and a dilution series (1, 2 and 3 μg) of total leaf protein extracted from transplastomic line 1. A dilution series of baculovirus-derived purified L1 VLPs (2, 4 and 8 ng) was used as reference. Other bands, in addition to that of GFP14-L1, were also observed, probably due to protein degradation. The amount of total recombinant protein was estimated to be up to 1.5% of TSP, as shown in Fig. 3c.
Chloroplast-derived L1 protein self assembles to capsomeres
Western blot analysis using monoclonal antibody MD2H11 and antigen capture ELISA with conformation-specific monoclonal antibody Ritti01 revealed the successful expression of L1 protein in all three transgenic lines. These data suggested that recombinant protein was present in higher order molecular structures. To verify these structures, cleared plant extracts were sedimented through a sucrose cushion followed by a CsCl density gradient centrifugation. L1 protein was analyzed in collected fractions (14 in total, fraction 1 from the bottom of the tube) by ELISA (Fig. 4a) using conformation-specific monoclonal antibody Ritti01. Most of the L1 protein was concentrated in fractions 6–10 as expected. Western blot analysis of these fractions also detected the GFP14-L1 protein (56.5 kDa) using HPV-16 L1-specific antibody MD2H11, as shown in Fig. 4b. These results suggested that chloroplast-derived L1 protein displayed conformational epitopes and assembled into higher order structures, such as capsomeres or VLPs.

Purification of chloroplast-derived L1 protein and the analysis of different assembly forms. a ELISA analysis of purified protein fractions obtained from CsCl density gradient centrifugation. Conformation-specific monoclonal antibody Ritti01 was the primary antibody for the detection of conformational epitopes. PBS was the control used for each collected protein fraction (lanes 1–14; fraction 1 corresponds to the tube bottom). b Western blot of CsCl fractions using MD2H11 monoclonal L1-specific antibody. The tobacco wild type was used as the control. c ELISA of 20 fractions obtained from sucrose gradient sedimentation (fraction 1 corresponds to tube bottom) using conformation-specific monoclonal antibody Ritti01. PBS served as the negative control for each fraction. Broken line Refractive index of the different fractions, arrow direction of sedimentation. d Western blot of the sucrose gradient sedimentation fractions using HPV-16 L1-specific monoclonal antibody MD2H11 for the detection of linear epitopes
To determine different assembly forms and to verify the assembly of L1 protein to capsomeres, we subjected crude extracts of tobacco leaves to sucrose sedimentation analysis. Among the 20 fractions collected (fraction 1 is from the bottom of the tube), capsomeres were expected in last fractions due to their low sedimentation coefficient. An ELISA of the sucrose sedimentation samples, using conformation-specific monoclonal antibody Ritti01, showed that most of the L1 protein was indeed concentrated in the final fractions (18–20; Fig. 4c), indicating conformational epitopes. The presence of a larger portion of L1 protein with a low sedimentation coefficient in the last fractions verified that most of the L1 protein assembled into capsomeres only. However, the detection of L1 protein in fractions 15–16 suggested the formation of other higher order structures. Western blot analysis by MD2H11 antibody further confirmed the presence of recombinant protein—mainly in fractions 18–20 (Fig. 4d). These data indicated that approximately 60% of the chloroplast-derived L1 protein assembled into capsomeres, as expected, while 40% of the L1 protein assembled into higher forms. In addition, some small amount of remaining unassembled L1 protein could still be present in the top fraction.
Discussion
We report here the possibility of producing a potentially cost-effective second-generation vaccine in tobacco chloroplasts through the expression of a mutated L1 gene (L1_2xCysM) that retains the assembly of L1 protein to capsomeres. The results presented here demonstrate that a reasonable amount of TSP (up to 1.5%) was obtained from the transplastomic tobacco plants. We also show that the correct formation of capsomeres was maintained, confirming their proper assembly by CsCl gradient centrifugation, sucrose gradient sedimentation analysis and ELISA with a conformation-specific monoclonal antibody. The expression of the L1_2xCysM gene appeared to induce male sterility in the plants, while rest of growth and morphological characteristics were observed to be normal.
There have been many arguments on the likelihood of being able to use VLP-based vaccines in developing countries due to their high cost, requirement of a cold chain and the necessity of sterile needles for intramuscular administration (Stanley et al. 2008). Keeping in mind that more than 80% of the cervical cancer cases occur in developing countries (Parkin and Bray 2006), there is a great need for a second-generation vaccine that is inexpensive to produce and thermo-stable (to circumvent cold chain) (Stanley et al. 2008), for these very properties would increase the availability of such a vaccine to developing countries. One possible alternative can be the capsomeres, which are considered to be relatively stable at room temperature and can be produced from Escherichia coli in large amounts (Li et al. 1997; McCarthy et al. 1998; Chen et al. 2001). As such, capsomeres represent an advantageous alternative to VLP-based vaccines for developing countries. However, there is still the question of cost-effectiveness as production of capsomeres from E. coli requires fermenters and the purification of protein (Daniell et al. 2009). This problem can be solved by choosing plants as the expression system: on the one hand, plants can be used for large-scale production, and on the other hand, plants can be ingested directly as a source of oral vaccines (Daniell et al. 2001).
In the study reported here, we opted chloroplast-based expression of a mutated L1 gene (L1_2xCysM), which due to the replacement of two highly conserved cysteine residues retains the assembly of L1 protein to capsomeres. The L1_2xCysM gene showed significant expression, with TSP estimated to be up to 1.5% in tobacco leaves. We used N-terminal fusion of the downstream box (DB), which consists of the first 14 amino acids of GFP (GFP14); this fragment has been reported to accumulate to high levels in plastids (Sidorov et al. 1999). The significant expression of L1 protein can either be due the protection of recombinant protein from degradation due to N-terminal fusion of DB or to an increased rate of translation, as indicated by Ye et al. (2001). Similar results were obtained when the L1 gene was linked to the DB consisting of a nucleotide sequence encoding the first 14 amino acids of Rubisco large subunit (rbcL) (Lenzi et al. 2008). Our results illustrate that the fusion of GFP14 to the L1_2xCysM gene did not affect the native conformation of L1 protein, as shown by the binding of L1 protein with conformation-specific antibody in ELISA.
The L1 gene has been expressed in tobacco plants following nuclear transformation, but only a very low protein accumulation was achieved (Biemelt et al. 2003; Varsani et al. 2003; Liu et al. 2005). However, Maclean et al. (2007) achieved a high yield (up to 11% of TSP) by targeting human codon-optimized L1 protein to the chloroplast. This increased yield may be due to a different protein hydrolyzing machinery in the plastids as well as to different protein stability conditions as a result of the presence of protective chaperones. In two recent studies, HPV-16 L1 protein-forming VLPs were expressed in the tobacco chloroplast, achieving 24% (Fernandez-San Millan et al. 2008) and 1.5% (Lenzi et al. 2008) of TSP, respectively. In contrast to our approach, Lenzi et al. (2008) used native viral L1 and a synthetic codon-optimized L1 gene. Lenzi et al. (2008) compared various constructs and found that L1 was only detectable when the N-terminus of the L1 protein was translationally fused with the first 14 amino acids of the N-terminal domain of the ATPase beta subunit (atpB) or the Rubisco large subunit (rbcL). Fernandez-San Millan et al. (2008) suggested that the fairly high accumulation of L1 protein achieved in their system may have been due to the use of the light-regulated psbA 5′-UTR.
All three transplastomic lines obtained in our study were found to be sterile. Although the plants produced flowers, these were either shed before maturity or they did not produce seeds. Seeds were obtained when the transplastomic lines were pollinated with wild-type tobacco plants, thereby demonstrating that sterility was restricted to the male organs only. However, in all transplastomic lines, the morphological characteristics of the vegetative organs were identical to those of wild-type plants. Subsequent studies on the T1 and T2 generations revealed the persistence of sterility in the transplastomic lines. Various factors can account for this persistence of sterility, such as the interference of novel open reading frames (ORFs) with metabolism in the cytoplasm, possibly due to lower levels of ATP production (Chase 2007; Pelletier and Budar 2007), or somaclonal variation induced during tissue culture (Larkin and Scowcroft 1981; Jain 2001). Male sterility in transplastomic tobacco plants was detected by Lössl et al. (2003), where the expression of poylydroxybutyrate (PHB) in tobacco chloroplasts resulted in male-sterile phenotypes. This effect was subsequently studied in detail by Ruiz and Daniell (2005) who found that the male-sterile phenotype had shortened stamens and produced nonviable pollen. These researchers attributed this effect to an alteration in chloroplast fatty acid metabolism due to the expression of phaA gene. A detailed study on the development and morphology of the male organs and backcrosses of the sterile phenotypes for several rounds would reveal the reasons for male sterility reported here. However, male sterility is advantageous as it facilitates hybrid seed production (Chase 2007). Additionally, male sterility can be used as a tool for further transgene containment by reducing the out-crossing risk of very small fraction of paternally transmitted plastids (Ruiz and Daniell 2005, Ruf et al. 2007).
We also investigated the proper assembly of L1 protein to higher order structures by CsCl density centrifugation. In this method, the particles move through the sucrose phase and are caught in the inter-phase between the sucrose and CsCl layer. As expected, L1 protein was detected in fractions 6–10 by western blot analysis. These fractions also immunoreacted with the conformation-specific monoclonal antibody Ritti01 in the ELISA, thereby indicating the presence of L1 protein in the form of higher order structures, such as capsomeres or VLPs. To verify that the expression of the L1_2xCysM gene actually did lead to the formation of capsomeres, we analyzed crude plant extracts by sucrose sedimentation analysis. This method allows quantitative separation of different assembly forms on the basis of their sedimentation coefficients. The ELISA and western blot analysis revealed that the L1 protein was largely concentrated in the final fractions (18–20), a result that confirmed that most of the L1 protein assembled into capsomeres with a low sedimentation coefficient. A second peak in ELISA was also observed in fractions 15–16, suggesting the formation of assembly forms other than capsomeres—ones with relatively higher sedimentation coefficients. Both CsCL density centrifugation and sucrose sedimentation analysis are standard techniques that have been previously used for the purification and quantitative separation of different assembly forms of plant-derived L1 protein by Biemelt et al. (2003), Maclean et al. (2007) and Fernandez-San Millan et al. (2008). All of these researchers reported two peaks pertaining to VLPs and capsomeres in the region of higher and lower sedimentation coefficients, respectively. However, with the modified L1 gene used in our study, VLP formation was unlikely because of the abolishment of cysteine residues. Consequently, the second peak observed in our sucrose gradient analysis may be due to small VLPs as it appeared in a sedimentation region between VLPs and capsomeres. In their sedimentation analysis of the modified L1 protein in which serines had replaced two cysteines, Schädlich et al. (2009b) observed the formation of capsomeres as well as higher assembly forms with higher sedimentation coefficients than capsomeres. In the same study, electron microscopy revealed that these higher assembly forms showing the sedimentation pattern related to small VLPs took the form of heterogeneous aggregates. The formation of heterogeneous rod-shaped structures for modified L1 protein having one cysteine replaced by serine at position 175 has also been reported (Ishii et al. 2003).
Many studies have focused on the immunogenicity of capsomeres that show the induction of neutralizing antibodies and T-cell responses (Rose et al. 1998; Fligge et al. 2001; Öhlschläger et al. 2003; Dell et al. 2006; Schädlich et al. 2009a). A promising approach towards capsomere-based vaccine was reported by Yuan et al. (2001) who demonstrated that dogs could be completely protected against papillomavirus infection by capsomeres. These researchers suggested that neutralizing epitopes do not necessarily need to be displayed in a complex, assembled structures (e.g., VLPs) for effective recognition by the host immune system. Rather, as suggested by Chen et al. (2000), such epitopes may be properly configured within the context of the pentameric capsomeres. Initially, in a direct comparison of different assembly forms (Thönes et al. 2008), VLPs were found to be more immunogenic than capsomeres. However, a recent study by Schädlich et al. (2009b) exemplified that capsomeres obtained by the expression of a mutated L1 gene induced antibody titres equivalent to those generated by VLPs. Although the mutated L1 gene (L1_2xCysM) used in our study is different from the mutated L1 gene reported to have higher immunogenicity by Schädlich et al. (2009b), it opens the door for the development of low-cost second-generation vaccine. Overall, these data imply that capsomeres are a promising candidate as a second-generation vaccine against HPV that can be affordable to developing countries, where cervical cancer is the leading cause of death among women (Parkin and Bray 2006).
Given the feasibility of chloroplast-derived VLP-based vaccines (Fernandez-San Millan et al. 2008; Lenzi et al. 2008) as well as the prospect of developing a cost-effective capsomere-based vaccine, we suggest that plastid expression of capsomeres is a valuable step towards the development of a second-generation vaccine against HPV.
References
Baker TS, Newcomb WW, Olson NH, Cowsert LM, Olson C, Brown JC (1991) Structures of bovine and human papillomaviruses. Biophys J 60:1445–1456
Biemelt S, Sonnewald U, Galmbacher P, Willmitzer L, Müller M (2003) Production of human papillomavirus type 16 viruslike particles in transgenic plants. J Virol 77:9211–9220
Bock R (2007) Plastid biotechnology: prospects for herbicide and insect resistance, metabolic engineering and molecular farming. Curr Opin Biotechnol 18:100–106
Chase CD (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet 23:81–90
Chebolu S, Daniell H (2009) Chloroplast-derived vaccine antigens and biopharmaceuticals: expression, folding, assembly and functionality. Curr Top Microbiol Immunol 332:33–54
Chen XS, Garcea RL, Goldberg I, Casini G, Harrison SC (2000) Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol Cell 5:557–567
Chen XS, Casini G, Harrison SC, Garcea RL (2001) Papillomavirus capsid protein expression in Escherichia coli: purification and assembly of HPV11 and HPV16 L1. J Mol Biol 307:173–182
Daniell H, Streatfield SJ, Wycoff K (2001) Medical molecular farming: production of antibodies, biopharmaceuticals and edible vaccines in plants. Trends Plant Sci 6:219–226
Daniell H, Sing ND, Mason H, Streatfield SJ (2009) Plant-made vaccine antigens and biopharmaceuticals. Trends Plant Sci 14:669–679
Dell K, Koesters R, Linnebacher M, Klein C, Gissmann L (2006) Intranasal immunization with human papillomavirus type 16 capsomeres in the presence of non-toxic cholera toxin-based adjuvants elicits increased vaginal immunoglobulin levels. Vaccine 24:2238–2247
Fernandez-San Millan A, Ortigosa SM, Hervas-Stubbs S, Corral-Martınez P, Seguı-Simarro JM, Gaetan J, Coursaget P, Veramendi J (2008) Human papillomavirus L1 protein expressed in tobacco chloroplasts self-assembles into virus-like particles that are highly immunogenic. Plant Biotechnol J 6:427–441
Fischer R, Stoger E, Schillberg S, Christou P, Twyman RM (2004) Plant-based production of biopharmaceuticals. Curr Opin Plant Biol 7:152–158
Fligge C, Giroglou T, Streeck RE, Sapp M (2001) Induction of type-specific neutralizing antibodies by capsomeres of human papillomavirus type 33. Virology 283:353–357
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158
Hajdukiewicz PTJ, Allison LA, Maliga P (1997) The two plastid RNA polymerases encoded by the nuclear and plastid compartments transcribe distinct groups of genes in tobacco. EMBO J 16:4041–4048
Harper DM, Franco EL, Wheeler C, Ferris DG, Jenkins D, Schuind A, Zahaf T, Innis B, Naud P, De Carvalho NS, Roteli-Martins CM, Teixeira J, Blatter MM, Korn AP, Quint W, Dubin G, GlaxoSmithKline HPV Vaccine Study Group (2004) Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomised controlled trial. Lancet 364:1757–1765
Ishii Y, Tanaka K, Kanda T (2003) Mutational analysis of human papillomavirus type 16 major capsid protein L1: the cysteines affecting the intermolecular bonding and structure of L1-capsids. Virology 308:128–136
Jain SM (2001) Tissue culture-derived variation in crop improvement. Euphytica 118:153–166
Koop HU, Herz S, Golds TJ, Nickelsen J (2007) The genetic transformation of plastids. Top Curr Genet 19:457–510
Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB, Chiacchierini LM, Jansen KU (2002) A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med 347:1645–1651
Larkin PJ, Scowcroft WR (1981) Somaclonal variation: a novel source of variability from cell cultures for plant improvement. Theor Appl Genet 60:197–214
Lenzi P, Scotti N, Alagna F, Tornesello ML, Pompa A, Vitale A, De Stradis A, Monti L, Grillo S, Buonaguro FM, Maliga P, Cardi T (2008) Translational fusion of chloroplast-expressed human papillomavirus type 16 L1 capsid protein enhances antigen accumulation in transplastomic tobacco. Transgenic Res 17:1091–1102
Li M, Cripe TP, Estes PA, Lyon MK, Rose RC, Garcea RL (1997) Expression of the human papillomavirus type 11 L1 capsid protein in Escherichia coli: characterization of protein domains involved in DNA binding and capsid assembly. J Virol 71:2988–2995
Liu HL, Li WS, Lei T, Zheng J, Zhang Z, Yan XF, Wang ZZ, Wang YL, Si LS (2005) Expression of human papillomavirus type 16 L1 protein in transgenic tobacco plants. Acta Biochim Biophys Sin (Shanghai) 37:153–158
Lössl A, Eibl C, Harloff HJ, Jung C, Koop HU (2003) Polyester synthesis in transplastomic tobacco (Nicotiana tabacum L.): significant contents of polyhydroxybutyrate are associated with growth reduction. Plant Cell Rep 21:891–899
Lössl A, Bohmert K, Harloff H, Eibl C, Mühlbauer S, Koop HU (2005) Inducible trans-activation of plastid transgenes: expression of the R. eutrophaphb operon in transplastomic tobacco. Plant Cell Physiol 46:1462–1471
Ma JKC, Barros E, Bock R, Christou P, Dale PJ, Dix PJ, Fischer R, Irwin J, Mahoney R, Pezzotti M, Schillberg S, Sparrow P, Stoger E, Twyman RM (2005) Molecular farming for new drugs and vaccines. Current perspectives on the production of pharmaceuticals in transgenic plants. EMBO Rep 6:593–599
Maclean J, Koekemoer M, Olivier AJ, Stewart D, Hitzeroth II, Rademacher T, Fischer R, Williamson AL, Rybicki EP (2007) Optimization of human papillomavirus type 16 (HPV-16) L1 expression in plants: comparison of the suitability of different HPV-16 L1 gene variants and different cell-compartment localization. J Gen Virol 88:1460–1469
Maliga P (2002) Engineering the plastid genome of higher plants. Curr Opin Plant Biol 5:164–172
McCarthy MP, White WI, Palmer-Hill F, Koenig S, Suzich JA (1998) Quantitative disassembly and reassembly of human papillomavirus type 11 viruslike particles in vitro. J Virol 72:32–41
Müller M, Gissmann L (2007) A long way: history of the prophylactic papillomavirus vaccine. Dis Markers 23:331–336
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Murray SL, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Oey M, Lohse M, Kreikemeyer B, Bock R (2009) Exhaustion of the chloroplast protein synthesis capacity by massive expression of a highly stable protein antibiotic. Plant J 57:436–445
Öhlschläger P, Osen W, Dell K, Faath S, Garcea RL, Jochmus I, Müller M, Pawlita M, Schäfer K, Sehr P, Staib C, Sutter G, Gissmann L (2003) Human papillomavirus type 16 L1 capsomeres induce L1-specific cytotoxic T lymphocytes and tumor regression in C57BL/6 mice. J Virol 77:4635–4645
Parkin DM, Bray F (2006) Chapter 2: the burden of HPV-related cancers. Vaccine 24S3:S11–S25
Pelletier G, Budar F (2007) The molecular biology of cytoplasmically inherited male sterility and prospects for its engineering. Curr Opin Biotechnol 18:121–125
Rose RC, White WI, Li M, Suzich JA, Lane C, Garcea RL (1998) Human papillomavirus type 11 recombinant L1 capsomeres induce virus neutralizing antibodies. J Virol 72:6151–6154
Ruf S, Karcher D, Bock R (2007) Determining the transgene containment level provided by chloroplast transformation. Proc Natl Acad Sci USA 104:6998–7002
Ruiz ON, Daniell H (2005) Engineering cytoplasmic male sterility via the chloroplast genome by expression of ß-ketothiolase. Plant Physiol 138:1232–1246
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sapp MC, Volpers C, Müller M, Streeck RE (1995) Organization of the major and minor capsid proteins in human papillomavirus type 33 virus-like particles. J Gen Virol 76:2407–2412
Sapp M, Fligge C, Petzak I, Harris JR, Streeck RE (1998) Papillomavirus assembly requires trimerization of the major capsid protein by disulfides between two highly conserved cysteines. J Virol 72:6186–6189
Schädlich L, Senger T, Kirschning CJ, Müller M, Gissmann L (2009a) Refining HPV 16 L1 purification from E. coli: reducing endotoxin contaminations and their impact on immunogenicity. Vaccine 27:1511–1522
Schädlich L, Senger T, Gerlach B, Mücke N, Klein C, Bravo IG, Müller M, Gissmann L (2009b) Analysis of modified human papillomavirus type 16 L1 capsomeres: the ability to assemble into larger particles correlates with higher immunogenicity. J Virol 83:7690–7705
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimada H, Sugiura M (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5:2043–2049
Sidorov VA, Kasten D, Pang SZ, Hajdukiewicz PTJ, Staub JM, Nehra N (1999) Stable chloroplast transformation in potato: use of green fluorescent protein as a plastid marker. Plant J 19:209–216
Smith JS, Lindsay L, Hoots B, Keys J, Franceschi S, Winer R, Clifford GM (2007) Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer 121:621–632
Stanley M, Gissmann L, Nardelli-Haefliger D (2008) Immunobiology of human papillomavirus infection and vaccination—implications for second generation vaccines. Vaccine 26S:K62–K67
Studier FW, Rosenberg AH, Dunn J, Dubendorf JW (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Meth Enzymol 185:60–89
Svab Z, Maliga P (1993) High-frequency plastid transformation in tobacco by selection for a chimeric aadA gene. Proc Natl Acad Sci USA 90:913–917
Svab Z, Hajdukiewicz P, Maliga P (1990) Stable transformation of plastids in higher plants. Proc Natl Acad Sci USA 87:8526–8530
Thönes N, Müller M (2007) Oral immunization with different assembly forms of the HPV 16 major capsid protein L1 induces neutralizing antibodies and cytotoxic T-lymphocytes. Virology 369:375–388
Thönes N, Herreiner A, Schädlich L, Piuko K, Müller M (2008) A direct comparison of human papillomavirus type 16 L1 particles reveals a lower immunogenicity of capsomeres than viruslike particles with respect to the induced antibody response. J Virol 82:5472–5485
Varsani A, Williamson AL, Rose RC, Jaffer M, Rybicki EP (2003) Expression of human papillomavirus type 16 major capsid protein in transgenic Nicotiana tabacum cv. Xanthi. Arch Virol 148:1771–1786
Villa LL, Costa RLR, Petta CA, Andrade RP, Paavonen J, Iversen OE, Olsson SE, Hoye J, Steinwall M, Riis-Johannessen G, Andersson-Ellstrom A, Elfgren K, Von Krogh G, Lehtinen M, Malm C, Tamms GM, Giacoletti K, Lupinacci L, Railkar R, Taddeo FJ, Bryan J, Esser MT, Sings HL, Saah AJ, Barr E (2006) High sustained efficacy of a prophylactic quadrivalent human papillomavirus types 6/11/16/18 L1 virus-like particle vaccine through 5 years of follow-up. Br J Cancer 95:1459–1466
Wakasugi T, Sugita M, Tsudzuki T, Sugiura M (1998) Updated gene map of tobacco chloroplast DNA. Plant Mol Biol Rep 16:231–241
Ye GN, Hajdukiewic PT, Broyles D, Rodriguez D, Xu CW, Nehra N, Staub JM (2001) Plastid-expressed 5-enolpyruvylshikimate-3-phosphate synthase genes provide high level glyphosate tolerance in tobacco. Plant J 25:261–270
Yuan H, Estes PA, Chen Y, Newsome J, Olcese VA, Garcea RL, Schlegel R (2001) Immunization with a pentameric L1 fusion protein protects against papillomavirus infection. J Virol 75:7848–7853
Yukawa M, Tsudzuki T, Sugiura M (2005) The 2005 version of the chloroplast DNA sequence from tobacco (Nicotiana tabacum). Plant Mol Biol Rep 23:359–365
Acknowledgments
A part of this work was conducted at Deutsches Krebsforschungszentrum, Germany. The study was partially financed by the GoF foundation. We want to express our gratitude for the support of Stefan Kirchner and Hans-Ulrich Koop at the Ludwig-Maximilians-University, Munich, Germany. Furthermore, we acknowledge the assistance of Elisabeth Geiger during the research work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Waheed, M.T., Thönes, N., Müller, M. et al. Transplastomic expression of a modified human papillomavirus L1 protein leading to the assembly of capsomeres in tobacco: a step towards cost-effective second-generation vaccines. Transgenic Res 20, 271–282 (2011). https://doi.org/10.1007/s11248-010-9415-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-010-9415-4