
Abstract
The catalytic domain of Acidothermus cellulolyticus thermostable endoglucanase gene (encoding for endo-1,4-β-glucanase enzyme or E1) was constitutively expressed in rice. Molecular analyses of T1 plants confirmed presence and expression of the transgene. The amount of E1 enzyme accounted for up to 4.9% of the plant total soluble proteins, and its accumulation had no apparent deleterious effects on plant growth and development. Approximately 22 and 30% of the cellulose of the Ammonia Fiber Explosion (AFEX)-pretreated rice and maize biomass respectively was converted into glucose using rice E1 heterologous enzyme. As rice is the major food crop of the world with minimal use for its straw, our results suggest a successful strategy for producing biologically active hydrolysis enzymes in rice to help generate alcohol fuel, by substituting the wasteful and polluting practice of rice straw burning with an environmentally friendly technology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The fuel ethanol industry has been growing extensively worldwide including the United States, Brazil and European Union countries (Ragauskas et al. 2006), and considerable efforts have been exerted towards improving ethanol yield and reducing its production costs during the last two decades (Lynd et al. 2005). In 2004, U.S. ethanol production capacity reached 3.4 billion gallons (Farrell et al. 2006). Another production increase of more than 1.6 billion gallons is projected for 2012 (Bothast and Schlicher 2005). The economic and environmental performance of maize seed and sugarcane ethanol produced in the U.S. and Brazil (Oliveira et al. 2005) would likely be improved by producing ethanol from lignocellulosic materials. Worldwide, over 1.7 billion tons of crop residues are available annually and nearly half of this total is rice straw (Kim and Dale 2004) which could mostly be used for processing to fuels and chemicals.
For ethanol to be produced from plant biomass sources, enzymatic hydrolysis of cellulose to fermentable sugars is employed (Kabel et al. 2005) using microbial hydrolysis enzymes (Ragauskas et al. 2006). These enzymes are produced in large-scale fermentation tanks (Knauf and Moniruzzaman 2004). Although the cost of enzyme production was reduced by about a factor of four from 1980 to 1999 (Wyman 1999) and by another 10 fold since 2000 (Knauf and Moniruzzaman 2004), it still represents about $0.30–0.50 per gallon of ethanol produced—a major cost factor (Bothast and Schlicher 2005). However, cost reduction might be achieved by producing crops that can sustainably and actively self-produce the desired hydrolysis enzymes (Ragauskas et al. 2006). One set of high value hydrolysis enzymes that might be alternatively produced within the biomass crops and utilized in fuel ethanol production in a biorefinery are the cellulases—enzymes that convert cellulose into fermentable sugars.
The E1 1,4-β-endoglucanase enzyme of Acidothermus cellulolyticus is one of the most thermostable cellulases known (Dai et al. 2005). The expression of the catalytic domain (cd) of the E1 1,4-β-endoglucanase in tobacco (Ziegelhoffer et al. 2001), potato (Dai et al. 2000), and Arabidopsis (Ziegler et al. 2000) demonstrated the possibility of producing this enzyme in plants. Several prominent crops have been recommended for this purpose, especially those with a high lignocellulosic biomass, some of which presently cause disposal problems (Kim and Dale 2004; Knauf and Moniruzzaman 2004).
Rice, as the primary source of caloric intake for over half of the world’s human population, is grown on over 148 million hectares worldwide (Chandra Babu et al. 2003) with a total production of about 731 million tons of straw (Kim and Dale 2004). While rice seed is traditionally the useful portion of this important crop, its remaining biomass has to date shown limited use. Customarily, farmers throughout the world burn rice straw in the field after harvest. Burning is inexpensive and mitigates rice diseases. However, emitted smoke gives rise to many health concerns such as increased incidence of asthma (Kayaba et al. 2004). These concerns, for example, resulted in California legislation that limits rice straw burning to the lesser of 25% of rice area in 2001, and even then burning is allowed only if evidence of disease is present (California Rice Commission 2005). As a result, California produces an excess of one million tons of rice straw each year. Of this produced amount, only about 3–4% is used in commercial applications and the rest must be incorporated into the soil which is a very costly process. Therefore, rice may become a viable bio-based energy candidate with potential to lower pollution levels at the same time. Moreover, the production of enzymes in rice straw may prove fruitful for the manufacture of other valuable bio-based industrial enzymes and protein pharmaceuticals in this crop.
This study examines the production of biologically active A. cellulolyticus endo-1,4-β-glucanase E1 enzyme in transgenic rice, conversion of crop biomass cellulose to glucose using the produced enzyme and the economic feasibility of producing this enzyme in rice.
Materials and methods
Transformation vector
The pZM766-E1cat containing the Acidothermus cellulolyticus E1 catalytic domain (cat) driven by the Cauliflower Mosaic Virus 35S Promoter (CaMV 35S), Tobacco Mosaic Virus Translational Enhancer (Ω), and the tobacco pathogenesis-related protein 1a (Pr1a) signal peptide encoding sequence for apoplast targeting and storage of the E1 enzyme was removed from the pUC19 vector by digestion with XbaI. The removed cassette was transferred to the binary vector pCAMBIA 3301 containing the bar herbicide resistance selectable marker and the gusA marker genes to generate the binary vector ApoE1.
Recovery of transgenic plants
Rice (Oryza sativa L. Japonica cv. Taipei 309) embryogenic callus induction, Agrobacterium-mediated transformation, and regeneration of transgenic plants were performed following the procedure previously described by Ahmad et al. (2002) with some minor modifications. Selection using 15 mg/l glufosinate ammonium was applied throughout callus proliferation, regeneration, and plant growth and rooting. The gusA-reporter gene expression assays were used to monitor the efficiency of transformation as outlined by Jefferson et al. (1987).
Total soluble proteins extraction
Total soluble proteins (TSP) were extracted from leaf tissues as described by Ziegelhoffer et al. (2001). Briefly, 100 mg fresh leaf tissue was ground in the sodium acetate grinding buffer (50 mM sodium acetate pH 5.5, 100 mM NaCl, 10% v/v glycerol, 0.5 mM ethylenediaminetetraacetic acid (disodium salt), 1 mM phenylmethylsulfonyl fluoride, 1 mg/l aprotinin, 1 mg/l leupeptin, 1 mg/l pepstatin) at a ratio of 5 μl per mg of sample. Soluble extract was recovered from insoluble debris after centrifugation for 5 min. A solution of saturated ammonium sulfate was added to the extracts to achieve a final concentration of 2.7 M ammonium sulfate. After incubation on ice for 30 min, the resulting precipitate was recovered by centrifugation for 5 min. The ammonium sulfate pellet was resuspended in 5 μl of grinding buffer for each 2 μl of starting crude extract. Extracts were quantified using the Bradford method using a standard curve generated from bovine serum albumin (BSA) (Ziegelhoffer et al. 2001).
MUCase activity assay of heterologous E1 enzyme
E1 activity was assessed as described by Ziegelhoffer et al. (2001). Briefly, a series of soluble protein dilutions ranging from 10−1 to 10−6 were developed. In a 96-well plate, 10 μl samples (representing 1–100 ng total soluble protein) was mixed with 100 μl reaction buffer [50 mM sodium acetate pH 5.5, 100 mM NaCl, 0.5 mM 4-methylumbelliferyl β-d-cellobioside (MUC)]. The fluorophore 4-methylumbelliferone (MU), as the product of E1 hydrolization of the substrate MUC, was measured as follows. Plates were covered with adhesive lids and incubated at 65°C for 30 min. The reaction was stopped with the addition of the stop buffer (0.15 M glycine pH 10.0), and the fluorescence was read at 465 nm using SPECTRAmax M2 device (Molecular Devices Inc., Sunnyvale, CA) at an excitation wavelength of 360 nm. After subtracting background fluorescence contributed by tobacco and rice control extracts, activity of each sample was calculated using a standard curve representing 4–160 pmol MU and compared to the activity of pure E1 reported in Ziegelhoffer et al. (2001).
DNA isolation and Southern blot hybridization analysis
Confirmation of transgene integration into the plant genome, number of independent transgenic lines, and transgene copy numbers were performed by Southern blot hybridization using the E1-coding sequence as a probe. For Southern blots, 8 μg of genomic DNA was digested with BstX1 restriction enzyme, electrophoresed in 1.0% (w/v) agarose gel, transferred onto Hybond-N+ (Amersham-Pharmacia Biotech) membranes, and fixed with a UV crosslinker (Stratalinker UV Crosslinker 1800, Stratagene, CA) as recommended in the manufacturers’ instructions. The E1 gene-specific probe was generated using PCR amplification of the E1 gene to produce a 1.0-kb fragment. The amplified fragment was purified using the QIAquick kit (QIAGEN). Probe labeling and detection were obtained using the DIG High Prime DNA Labeling and Detection Starter Kit II (Kit for chemiluminescent detection with CSPD, Roche Co.), following the manufacturer’s protocol.
RNA isolation and Northern blot hybridization analysis
Total RNA samples of untransformed and transgenic plants were isolated from five different transgenic rice lines using the TRI Reagent (Sigma-Aldrich, St. Louis, Mo) according to the manufacturer’s instructions. Also, RNA samples were extracted from tobacco plants transformed with the same gene and used as a positive control for comparison in this study (Ziegelhoffer et al. 2001). Aliquots of RNA (20 μg) were fractionated in 1.2% agarose formaldehyde denaturing gel and blotted on a Hybond-N+ nylon membrane (Amersham Pharmatica Biotech) as specified by the manufacturer. The E1 gene-specific probe was generated using PCR amplification of the E1 gene to produce a 1.0-kb fragment. The fragment was gel purified using the QIAquick Gel Extraction Kit (QIAGEN Inc., Valencia, CA). Probe labeling and transcript detection were obtained using the DIG High Prime DNA Labeling and Detection Starter Kit II (Kit for chemiluminescent detection with CSPD, Roche Co.), following the manufacturer’s protocol.
Western blot analysis
Invitrogen NuPAGE® Bis-Tris Discontinuous Buffer System with the 10% NuPAGE® Novex Bis-Tris Pre-Cast Gel was used. Total soluble protein (1 μg), NuPAGE® LDS Sample Buffer (5 μl), NuPAGE® Reducing Agent (2 μl), and deionized water were mixed to a total volume of 20 μl. The samples were heated at 70°C for 10 min prior to electrophoresis using the XCell SureLockTM Mini-Cell with NuPage® MES SDS Running Buffer. The gel was run for about 45 min at 200 V, and blotted onto a membrane using the XCell II® Blot Module and NuPAGE® Transfer Buffer at 30 V for 1 h, following the manufacturer’s protocol. The membrane was placed into blocking buffer (1× PBS, 5% non-fat dry milk, and 0.1% Tween 20) immediately after transfer and incubated at room temperature for 1 h with gentle agitation. The primary antibody (mouse anti-E1, provided by Steven Thomas, National Renewable Energy Laboratories) was diluted in blocking buffer to a concentration of 1 μg/ml. The blocking buffer was decanted from the membrane, 10 ml of antibody solution was added, and the membrane was incubated at room temperature for 1 h with gentle agitation. The primary antibody solution was decanted and the membrane was washed in washing buffer (1× PBS, 0.1% Tween 20) for 30 min with gentle agitation at room temperature, changing the wash solution every 5 min. The enzyme conjugate anti-mouse IgG:HRPO (Transduction Laboratories) was diluted 1:2000 in blocking solution and added to the membrane after decanting the wash buffer. The membrane was incubated with the secondary antibody solution for 1 h at room temperature with gentle agitation; the antibody solution was decanted from the membrane and the membrane was washed in washing solution as before. For detection, 1 ml each of Stable Peroxide Solution and Luminol/Enhancer Solution (Pierce SuperSignal® West Pico Chemiluminescent Substrate) were mixed and incubated with the membrane for 5 min. The membrane was blotted slightly to remove excess substrate and placed in a plastic envelope. Excess liquid and air bubbles were removed. The blot was exposed to X-ray film (Kodak BioMax XAR Scientific Imaging Film) and developed in a Kodak RP X-OMAT Processor.
Immunofluorescence microscopic analysis
Free-hand sections of fresh leaf tissue from transgenic and untransformed rice plants were isolated and hydrated in NaCl/Pi buffer (0.8% NaCl, 0.02% KCl, 0.14% Na2HPO4 · 2H2O, and 0.02% KH2PO4 in water) containing 0.5% BSA (BSA/NaCl/Pi) for 2 min. Sections were incubated in primary antibody [rabbit anti-(mouse IgG)] raised against the E1 enzyme diluted 1:250 in the same buffer, in a moist chamber for 3 h. The primary antibody was rinsed off with the BSA/NaCl/Pi buffer and sections were incubated for 2 h at room temperature with fluorescein isothiocyanate (FITC)-conjugated secondary antibody [goat anti-(rabbit whole molecule IgG)] diluted 1:250 in the same buffer using same moist chamber. The secondary antibody was then rinsed off with the same buffer. Intracellular localization of the FITC-labeled protein was observed and images were taken using a confocal laser scanning microscopy Zeiss LSM 5 Pascal (Carl Zeiss, Jena, Germany). FITC fluorescence and chloroplast autofluorescence was excited with an argon ion laser, λex = 488 nm. Fluorescence emission was detected through a Band Pass (BP) filter, λem = 530/30 nm for the FITC (images represented in green) and Long Pass (LP) filter, λem = 650 nm for the chloroplast (images represented in red). Either a 63× Plan-apochromat or a 20× Plan-neofluar objective lens was used.
Cellulose hydrolysis assay
Milled corn stover and rice straw (about 1 cm in length) were pretreated using Ammonia Fiber Explosion technique (AFEX). The biomass was transferred to a high pressure Parr reactor with 60% moisture (kg water/kg dry biomass) and liquid ammonia ratio 1.0 (kg of ammonia/kg of dry biomass) was added. As the temperature was slowly raised, the pressure in the vessel increased. The temperature was maintained at 90°C for five minutes before explosively releasing the pressure. The instantaneous drop of pressure in the vessel caused the ammonia to vaporize, causing an explosive decompression and considerable fiber disruption. The pretreated material was kept under a hood to remove residual ammonia and stored in a freezer until further use.
E1 activity was measured by reacting total protein extracted from E1-expressed rice leaves with different substrates, namely: AFEX-treated corn stover (CS), AFEX-treated rice straw (RS), CMC and Avicel. Commercial cellulase enzyme (15 FPU/g glucan; 31.3 mg/g glucan of Spezyme CP from Genencor) was used in this experiment as a control. The enzyme hydrolysis was done in a sealed scintillation vial. The substrates were hydrolyzed at a glucan loading of 1% (w:v) in a reaction medium composed of 7.5 ml of 0.1 M, pH 4.8 sodium citrate buffer was added to each vial. In addition, 60 μl (600 μg) tetracycline and 45 μl (450 μg) cycloheximide were added to prevent the growth of microorganisms during the hydrolysis reaction. The reaction was supplemented with 30 CBU of β-glycosidase enzyme (Novo 188 from Sigma) to avoid inhibition by cellobiose. Distilled water was then added to bring the total volume in each vial to 15 ml. Calculations were done based on theoretical glucan for corn stover (34.4%) and rice straw (45.2%). All the reactions were done in duplicate to test reproducibility. All hydrolysis reactions were carried out at 50°C with a shaker speed 90 rpm. About 1 ml of sample was collected at 72 and 168 h of hydrolysis, filtered using a 0.2 μm syringe filter and kept frozen.
Hydrolyzate was quantified using Waters HPLC by running the sample in Aminex HPX-87P (Biorad) column, against sugar standards. The amount of glucose produced in the enzyme blank and substrate blank were subtracted from the respective hydrolyzate glucose levels.
Results
Transgene construct and genetic transformation
The ApoE1 binary vector containing the catalytic domain (cat) of the A. cellulolyticus thermostable endoglucanase (E1) gene (encoding for 1,4-β-endoglucanase enzyme) was introduced into the nuclear genome of mature embryo-derived calli of the rice variety Taipei 309 (Oryza sativa L. subsp. Japonica) using the Agrobacterium-mediated transformation system (Ahmad et al. 2002). Callus transformation frequency, as defined in terms of percentage of glufosinate herbicide resistant calli was 32%. After being transferred onto regeneration media, about 78% of these transformants differentiated into plantlets in the presence of 15 mg/l glufosinate ammonium. The average transformation efficiency as defined in terms of number of independent transgenic lines to total number of calli inoculated was approximately 10%. Twenty-nine independent events were produced, among which the five transgenic events that showed the highest enzymatic activity were selected for further analysis. T0 and T1 plants grew well with no apparent growth or developmental abnormalities under controlled growth chamber and greenhouse environments.
High-level production of biologically active E1 enzyme
E1 enzyme was expressed at relatively high levels of 2.4–4.9% of leaf total soluble protein (LTSP), as detected among the rice transgenic lines. These results suggest that the microbial E1 enzyme remained biologically active in the transgenic rice plants while E1 activity was not present in the untransformed plants (Fig. 1).

Percentage of heterologous E1 enzyme in different independent transgenic rice events determined by the MUCase activity assay (average of 3 reps)
Molecular analyses of the transgenic plants
Combining the results of stable GUS expression patterns (for the gusA gene) and PCR (for the bar and E1 genes) assays confirmed the presence of intact, linked gusA, bar and E1 genes. The blue color of GUS expression patterns was observed in the transgenic calli and plantlets. The expected PCR bands for bar and E1 genes were confirmed in the plasmid as well as the transgenic rice lines, but not in the untransformed control plants (data not shown).
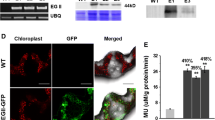
Southern blot analysis confirmed the stable incorporation, copy number and independence of the transgenic lines (Fig. 2a). The genomic DNA of the five transgenic lines showed bands of different sizes, as an indication of five independent transgenic events with 1–2 copies.

Southern (a), northern (b) and western (c) blot analyses show the presence of the transgenes in five transgenic rice lines (a) Southern blot analysis showing different integration patterns for the five transgenic rice lines. P: plasmid; C: non-transgenic; 1–5: transgenic lines. (b) Upper: northern blot analysis showing 1 kb bands for the five transgenic control; 1–5: transgenic lines. Lower: RNA loading control panel. (c) Western blot analysis showing 40 kDa bands for the five transgenic rice lines. +: positive control; C: non-transgenic control; 1–5: transgenic lines
Northern blot analysis was used to confirm the transcription of the E1 gene. A transcript of approximately 1 kb for this gene was detected in the tobacco positive control plants (Ziegelhoffer et al. 2001) as well as the rice transgenic lines. These results indicate that the transgenic lines possess the transcriptionally-active E1 gene (Fig. 2b).
Western blot analysis of leaf total soluble proteins (LTSP) using the mouse antibody raised against the E1 protein confirmed the expression of E1 both in transgenic tobacco and rice with the expected molecular mass of 40-kDa (Fig. 2c). Furthermore, the relative amount of transcript and 40 kDa E1 polypeptide in all five transgenic rice lines, judged from band intensity respectively in northern and western blots, correlated well with the amount of E1 enzyme produced in the transgenic lines using the 4-methylumbelliferyl β-d-cellobioside assay (MUCase) (Figs. 1 and 2b, c).
Localization of the E1 enzyme in the apoplast
Strong green fluorescent signals were detected in the apoplast of the transgenic tissues upon the application of immunoflouresence scanning laser confocal microscopy, confirming accumulation of the E1 enzyme. No signals were detected in the untransformed control plant tissues (Fig. 3).

Immunofluorescence confocal microscopy for the transgenic (a) and untransformed (b) rice showing apoplast localization of the E1 enzyme in transgenic rice leaves
Cellulose to glucose conversion
The Ammonia Fiber Explosion (AFEX)-pretreated (Teymouri et al. 2004) maize and rice biomass (lignocellulosic substrates containing both amorphous and crystalline cellulose), as well as increasing concentrations of both CMC (amorphous cellulose) and Avicel (crystalline cellulose) were converted into glucose using the transgenic rice plant total soluble proteins containing the E1 enzyme. Using 10% CMC and Avicel concentrations, approximately 0.6 and 0.25 g/l glucose were released after 168 h of hydrolysis, respectively (Fig. 4a). Additionally, considerable amounts of polyoligosaccharides were released from the CMC substrate blank, and the apparent solution viscosity increased substantially. Conversely, when an aliquot of rice E1-containing total soluble proteins was added to the CMC substrate, viscosity declined with reduced polyoligosaccharide formation and a detectable increase in the glucose peak (data not shown).

(a) The amount of glucose released from the enzymatic hydrolysis of CMC (1%, 5%, 10%) and Avicel (1%, 5%, 10%) using total protein extracted from E1 expressed rice straw. (b) Comparison of percentage of glucan converted in the enzymatic hydrolysis of corn stover (CS) and rice straw (RS). CE, commercial enzyme, UT, untreated biomass, CS1, RS1, CS2, and RS2 represent, reaction done using 0.5 ml and 4 ml of total soluble protein (with 4.9% of E1) and commercial β-glucosidase (6.5 mg/15 ml) respectively
Approximately 25% and 95% cellulose conversion was achieved for untreated and AFEX-treated corn stover respectively, when the cellulase commercial enzyme (Spezyme CO, Genencore) along with β-glucosidase (Novo 188, Sigma) was used in each case. Under similar conditions, untreated and AFEX-treated rice straw showed 21% and 62% cellulose conversion respectively. Since both untreated corn stover and rice straw showed much lower conversion compared to AFEX-treated biomass (less than 2% using E1-containg rice extract and 25% and 21% using cellulase commercial, respectively), AFEX-treated biomass was used for further experiments. When 0.5 ml of rice extract containing 4.9% LTSP E1 along with commercial β-glucosidase were added to the substrates, 17% and 14% of cellulose was respectively converted for AFEX-treated corn stover and AFEX-treated rice straw. When the amount of E1-bearing rice extract was increased to 4 ml, 30% and 22% were respectively converted under the same conditions. No activity was observed when substrates were treated with non-transgenic (NT) rice total soluble protein (Fig. 4b).
Discussion
Plants have been used as “green bioreactors” for the production of essential enzymes (Hong et al. 2004; Chiang et al. 2005) and other proteins (Liu et al. 2005), carbohydrates (Sahrawy et al. 2004) and lipids (Qi et al. 2004) while requiring minimal inputs of raw materials and energy (Teymouri et al. 2004). Production of biomolecules in plants, considered as molecular farming, is one approach to improve the economics and increase the low-cost production efficiency of these biomolecules (Fischer et al. 2004).
Several crops have been recommended for biomass-to-ethanol conversion, among them maize, rice, sugarcane and switchgrass (Kim and Dale 2004; Knauf and Moniruzzaman 2004)—all with a high amount of lignocellulosic biomass, and some of which have caused disposal problems. Production of enzymes in plants used for biomass conversion is a potentially powerful tool to facilitate the conversion of cellulose to glucose in the commercial production of ethanol while solving the problems associated with accumulated agricultural waste biomass. It is when ethanol bioconversion enzyme costs are decreased, that ethanol biorefineries achieve financial advantages over petroleum refineries.
When accumulated in cytosol, the normal level of heterologous protein production in plants is usually no more than 0.1–0.3% of plant total soluble proteins. The high level of E1 production in this research (Fig. 1) and in Arabidopsis (Ziegler et al. 2000) might be due to the fact that the E1 has been targeted to apoplast for accumulation. Among many other factors, the use of the catalytic domain of the E1 gene and the Tobacco Mosaic Virus translational enhancer, and the strength of the CaMV 35S promoter (Cheng et al. 2004) might have contributed to the overall level of production of E1 in rice.
It has been well documented that cellulases work together synergistically to decrystallize and hydrolyze the cellulose. Exo-glucanases act on crystalline cellulose (on cellulose chain ends) and endo-glucanase (E1) acts on amorphous cellulose (interior portions of the cellulose chain) (Bayer et al. 1998). In contrast, the results in this study demonstrate production of glucose from atypical endoglucanase (i.e. E1) activity on crystalline cellulose (Avicel). This could be because E1 enzyme can cause multiple random attacks on the same cellulose chain resulting in small fragments of cellobiose, cellotriose and cellotetraose. These fragments can be further hydrolyzed by enzyme molecules in solution such as E1 itself or the β-glycosidase enzyme used to avoid reaction inhibition by cellobiose (Medve et al. 1998).
Economic analysis
There are three hurdles to be cleared before endoglucanase-producing rice straw is available for commercial production. First, production of exoglucanase enzyme along with the endo-1,4-beta glucanase, would improve profitability. Second, if the rice grain is sold into the U.S. food chain, then the endoglucanases should not be present in the grain to avoid any adverse consumer reaction. Third, gene expression, optimizing downstream processing and purification of the endoglucanase from rice straw as well as biomass conversion reactions need to be perfected on a commercial basis.
Despite these hurdles, we proceed with an economic analysis of the benefits and costs of plant-based endoglucanase production, on the assumption that these technical issues can be solved. Two additional assumptions go into the construction of the analysis. First, we provide economic rather than financial analysis of the straw. Given that California, for example, is subsidizing the development of alternative uses for rice straw, the financial analysis should be more positive (or less negative) than the economic analysis. Second, we assume that there will be a market for large quantities of glucanase. This market will exist if current attempts to convert a significant proportion of U.S. automobiles to the gasoline E85 are successful.
The economic cost of producing rice straw as a by-product of grain production is the cost of harvesting, baling and transporting which is less than the cost of soil incorporation. According to CSU Chico, final report to California Air Resources Board on the development of a commercial scale composting plant in Colusa County, the cost of rice straw harvesting and baling is $16/metric ton. Transportation costs per ton are computed as a fixed cost of $5.50 plus a cost of $0.088 per mile. Thus, for a 25 mile haul the cost would be about $7.7/metric ton (Rice Straw Feedstock Joint Venture 1999). The exact transportation costs will depend on the location of the processing facility and the contract farms. Incorporating rice straw into the soil is estimated to be $43/acre and rice produces about 2.2 metric tons/acre of straw (California Rice Commission 2005). Combining these numbers leads to a cost of net delivered rice straw materials of $9.14/acre (Table 1).
Rice straw contains approximately 5% crude protein on a dry-matter basis, and 40% of this protein is soluble (NRC 1996). As demonstrated in the laboratory analysis, the endoglucanase production is 5% of the total soluble protein. These numbers result in an estimate of 2.2 kg of enzyme/acre (Table 1). As novel enzyme extraction from rice straw has not been accomplished, there is no information available on these figures. Therefore, we assume an approximation of $5/kg enzyme for the processing costs, at 95% recovery rate. This figure includes amortized fixed costs (plant, equipment etc.) plus variable costs (electricity, labor, etc.). These calculations lead to a $9.64 total cost of kg enzyme (Table 1).
The results and economic analysis of this study indicate that the production of glucanase enzymes in plant biomass might potentially be commercially viable, with the caveat that for this potential to be fulfilled additional work is needed to be able to produce the full range of commercial enzymes used in ethanol production and to provide continued technical improvements and cost reductions. In addition to the cost of raw material, the higher protein content requires less crop to be grown and harvested, which is important when considering the cost of crop handling, processing, and protein extraction (i.e., the less the amount of biomass per unit weight of recombinant protein, the less the extraction cost will be). Generally, it was estimated that the cost of producing 1 kg of recombinant protein from crops is 10–50 times lower than the cost of producing the same amount by E. coli fermentation (Kusnadi et al. 1997). This is true assuming that each crop could produce the target protein as 10% of the total soluble protein and the recombinant protein is 20% of total E. coli protein (Kusnadi et al. 1997).
It is also important to mention that because lignin is the biofuel-blocking polymer in biomass conversion (Sticklen 2006), one might also produce ligninases within the crop biomass similar to when the laccase was produced in maize seeds (Bailey et al. 2004) to reduce the needs for pretreatment processes (Sticklen 2006). When all these enzymes are produced at a very high level (Ziegler et al. 2000) in plants, this could well compete with the full range of commercial hydrolysis enzymes currently used in ethanol production. Furthermore, research is needed to ensure that lignocellulosic conversion enzymes produced within the plants will survive pretreatment, harvest, storage and transportation.
References
Ahmad A, Maqbool SB, Riazuddin S, Sticklen MB (2002) Expression of synthetic cry1ab and cry1ac genes in basmati rice (Oryza sativa L.) variety 370 via Agrobacterium-mediated transformation for the control of the European corn borer (Ostrinia nubilalis) In Vitro Cell Dev Biol 38:213–220
Bailey MR, Woodard SL, Callaway E, Beifuss K, Magallanes-Lundback M, Lane JR, Horn ME, Mallubhotla H, Delaney DD, Ward M, Van Gastel F, Howard JA, Hood EE (2004) Improved recovery of active recombinant laccase from maize seed. Appl Microbiol Biotechnol 63:390–397
Bayer EA, Chanzy H, Lamed R, Shoham Y (1998) Cellulose, cellulases and cellulosomes. Curr Opin In Str Biol 8:548–557
Bothast RJ, Schlicher MA (2005) Biotechnological processes for conversion of corn into ethanol. Appl Microbiol Biotechnol 67: 19–25
California Rice Commission (2005) Introduction to rice straw management. Available at http://www.calrice.org
Chandra Babu R, Nguyen BD, Varapong Chamarerk, Shanmugasundaram P, Chezhian P, Jeyaprakash P, Ganesh SK, Palchamy A, Sadasivam S, Sarkarung S, Wade LJ, Nguyen HT (2003) Genetic analysis of drought resistance in rice by molecular markers association between secondary traits and field performance. Crop Sci 43:1457–1469
Cheng M, Lowe BA, Spencer TM, Ye X, Armstrong CL (2004) Factors influencing Agrobacterium-mediated transformation of monocotyledonous species. In Vitro Cell Dev Biol 40:31–45
Chiang CM, Yeh FS, Huang LF, Tseng TH, Wang CS, Lur HS, Shaw JF, Yu SM (2005) Expression of a bifunctional and thermostable amylopullulanase in transgenic rice seeds leads to starch autohydrolysis and altered composition of starch. Mol Breed 15:125–143
Dai Z, Hooker BS, Anderson DB, Thomas SR (2000) Improved plant-based production of E1 endoglucanase using potato: expression optimization and tissue targeting. Mol Breed 6:277–285
Dai Z, Hooker BS, Quesenberry RD, Thomas SR (2005) Optimization of Acidothermus celluloyticus endoglucanase (E1) production in transgenic tobacco plants by transcriptional, post-transcription and post-translational modification. Transgenic Res 14:627–643
Farrell AE, Plevin RJ, Turner BT, Jones AD, O’Hare M, Kammen DM. (2006) Ethanol can contribute to energy and environmental goals. Science 311:506–508
Fischer R, Stoger E, Schillberg S, Christou P, Twyman R (2004) Plant-based production of biopharmaceuticals. Curr Opin Plant Biol 7:152–158
Hong CY, Chen KJ, Liu LF, Tseng TH, Wang CS, Yu SM (2004) Production of two highly active bacterial phytases with broad pH optima in germinating transgenic rice seeds. Trans Res 13:29–39
Jefferson RA, Kananagh TA, Bevan MW (1987) GUS fusions: Glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3301–3306
Kabel MA, Van der Maarel MJEC, Klip G, Voragen AGJ, Schols HA (2005) Standard assays do not predict the efficiency of commercial cellulase preparations towards plant materials. Biotechnol Bioeng 93:56–63
Kayaba H, Meguro H, Muto H, Kamada Y, Adachi T, Yamada Y, Kanda A, Yamaguchi K, Hamada K, Ueki S, Chihara J (2004) Activation of Eosinophils by rice-husk dust exposure: a possible mechanism for the aggravation of asthma during rice harvest. Tohoku J Exp Med 204:27–36
Kim S, Dale BE (2004) Global potential bioethanol production from wasted crops and crop residues. Biomass Bioenergy 26:361–375
Knauf M, Moniruzzaman M (2004) Lignocellulosic biomass processing: A perspective. Internat Sugar Jour 106:147–150
Kusnadi AR, Nikolov ZL, Howard JA (1997) Production of Recombinant Proteins in Transgenic Plants: Practical Considerations. Biotechnol Bioeng 56:473–484
Liu HL, Li WS, Lei T, Zheng J, Zhang Z, Yan XF, Wang ZZ, Wang YL, Si LS (2005) Expression of Human Papillomavirus type 16 L1 protein in transgenic tobacco plants. Acta Biochim Biophys Sin 37:153–158
Lynd LR, van Zyl WH, McBride JE, Laser M (2005) Consolidated bioprocessing of cellulosic biomass: an update. Curr Opin Biotechnol 16:577–583
Medve J, Karlsson J, Lee D, Tjerneld F (1998) Hydrolysis of microcrystalline cellulose by cellobiohydrolase I and endoglucanase II from Trichoderma reesei: Adsorption, sugar production pattern and synergism. Biotechnol Bioeng 59:621–634
NRC (1996) Nutrient requirements of beef cattle, 7th Revised edn. National Academy Press, Washington, DC
Oliveira ME, Vaughan BE, Rykiel EJ (2005) Ethanol as fuel: Energy, carbon dioxide balances, and ecological footprint. BioScience 55:593–602
Qi B, Fraser T, Mugford S, Dobson G, Sayanova O, Butler J, Napier JA, Stobart AK, Lazarus CM (2004) Production of very long chain polyunsaturated omega-3 and omega-6 fatty acids in plants. Bio/technology 22:739–745
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ Jr, Hallett JP, Leak DJ, Liotta CL et al (2006) The path forward for biofuels and biomaterials. Science 311:484–489
Rice Straw Feedstock Joint Venture (1999) Rice Straw Feedstock Supply Study for Colusa County California, Western Regional Biomass Energy Program, Lincoln, NE
Sahrawy M, Avila C, Chueca A, Canovas FM, Lopez-Gorge J (2004) Increased sucrose level and altered nitrogen metabolism in Arabidopsis thaliana transgenic plants expressing antisense chloroplastic fructose-1,6-bisphosphatase. J Exp Bot 55:2495–2503
Sticklen M (2006) Plant genetic engineering to improve biomass characterization for biofuels. Curr Opin Biotechnol 17:315–319
Teymouri F, Alizadeh H, Laureano-Perez L, Dale BE, Sticklen M (2004) Effects of ammonia fiber explosion treatment on activity of endoglucanase from Acidothermus cellulolyticus in transgenic plant. Appl Biochem Biotechnol 116:1183–1192
Wyman CE (1999) Biomass ethanol: Technical progress, opportunities, and commercial challenges. Annu Rev Energ Env 24:189–226
Ziegelhoffer TJ, Raasch A, Austin-Phillips S (2001) Dramatic effects of truncation and subcellular targeting on the accumulation of recombinant microbial cellulase in tobacco. Mol Breed 8:147–158
Ziegler MT, Thomas SR, Danna KJ (2000) Accumulation of a thermostable endo-1,4-β-d-glucanase in the apoplast of Arabidopsis thaliana leaves. Mol Breed 6:37–46
Acknowledgments
This work was supported by DOE/Edenspace (Cooperative Agreement # DE-FG02-04ER86183), Government of Egypt, MSU Research Excellence Funds (REF) and Consortium for Plant Biotechnology Research (CPBR Agreement # GO12026-168). The authors are thankful to National Renewable Energy Laboratory for providing the E1 antibodies, Dr. K. Danna for providing the pZM766-E1cat, USDA-ARS in Beaumont, Texas and the National Small Grains Collection at Aberdeen, Idaho for providing the rice seeds. The authors appreciate the critical review of the manuscript by Dr. Brian Hooker and Dr. Chris Somerville.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oraby, H., Venkatesh, B., Dale, B. et al. Enhanced conversion of plant biomass into glucose using transgenic rice-produced endoglucanase for cellulosic ethanol. Transgenic Res 16, 739–749 (2007). https://doi.org/10.1007/s11248-006-9064-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-006-9064-9