
Abstract
Hydrogenation of succinic acid aqueous solutions was performed using TiO2-supported 2 wt% Pd and 2 wt%Pd–x wt%Re catalysts, using either impregnation method or surface redox reduction of the monometallic catalyst. The former catalysts were superior in terms of activity and selectivity to 1,4-butanediol than the latter ones. However, higher Re loadings (3.4–3.6 wt% compared to 0.6–0.8 wt%) were necessary to initiate this synergy.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Succinic acid (SUC) is a platform molecule from which several high-volume and high-value derivative compounds can be synthesized [1–4]. These include 1,4-butanediol (BDO) an intermediate for other chemicals or polymers. One common industrial method of producing this diol is the maleic anhydride-based catalytic hydrogenation [5, 6]. There is extensive patent literature regarding the hydrogenation of maleic anhydride, acid or dialkylester from fossil origin to produce BDO, but also γ-butyrolactone (GBL) and tetrahydrofuran (THF), and the data are summarized in some reviews [7–9]. Analogously to maleic anhydride, SUC may be converted to GBL, BDO, and THF by catalytic hydrogenation (Scheme 1) [7–17].

Catalytic hydrogenation of succinic acid (SUC) to γ-butyrolactone (GBL), 1,4-butanediol (BDO) and tetrahydrofuran (THF)
SUC is also among the new bio-derived chemicals from renewable resources, such as starch or oligosaccharides (C-5, C-6 sugars) [1–4]. A lot of research has been performed in the past decade on the fermentative production of bio-SUC which is now produced at a lower cost than petro-derived SUC [8, 9, 18–20]. Several companies are currently involved in R&D, pilot plants and large scale production, such as BioAmber (ARD-France/DNPGreen Technology), Reverdia (DSM/Roquette), Myriant Technologies LCC, Mitsubishi Chemical Corporation/PTT Public Company Limited-Thailand, BASF/Purac-CSM, [21, 22].
Since after the fermentation process SUC is obtained in aqueous phase, heterogeneous catalysts that are able to hydrogenate SUC in aqueous phase must be designed [3, 9, 23]. However, little work has been developed on the direct aqueous phase catalytic conversion of SUC [3, 13, 23–25] and most of the publications in liquid phase use organic solvents (dioxane, aqueous ethanol) [11, 12, 14–17]. Moreover, it is known that it is very difficult to hydrogenate free carboxylic acids to the corresponding alcohols compared to the esters and this reaction requires severe conditions of temperature and pressure (>150 °C, >100 bar) [26]. Among the various heterogeneous hydrogenation catalysts examined for hydrogenation of maleic acid or succinic anhydride/acid, mainly in patents, metal-promoted noble metal supported catalysts have been reported to be effective in water [9, 25]. In our previous study dealing with the hydrogenation of an aqueous 15 wt% SUC aqueous solution at 160 °C under 150 bar, we reported that monometallic Pd/C catalysts were very selective to GBL, whereas Pd–Re/C catalysts prepared by impregnation of the monometallic catalyst with an aqueous solution of NH4ReO4 were very effective for the synthesis of BDO [13]. However, a relatively high Re/Pd weight ratio was necessary. The best BDO selectivity (66 %) was observed when ca. 4.0 wt% Re was added to the 2.0 wt% Pd/C catalyst.
In continuation of our previous work, the objective of this study was to evaluate titania supported catalysts. These materials are stable under hydrothermal conditions [27]. Also, compared to microporous active carbon developing usually high specific surface areas, these supports may adsorb fewer residual impurities present in the bio-SUC and that are potential poisons for the catalyst [28]. Pd–Re/TiO2 catalysts were prepared using two different methods of Re deposition which should lead to different distributions of Re sites on the Pd/TiO2 catalysts and were evaluated in the hydrogenation of an aqueous solution of SUC. The objective was also to try to minimize the loading amount of Re promoter to yield a catalyst selective to BDO.
2 Experimental Section
2.1 Catalysts Preparation and Characterization
Two commercial titania (Millenium DT51, specific area = 90 m2 g−1 and Degussa P25, specific area = 50 m2 g−1), noted TiO2 A and TiO2 B, respectively, were used as supports. Monometallic Pd/TiO2 catalysts were prepared via a deposition–precipitation method using an aqueous phase of potassium tetrachloropalladate, so as to obtain a palladium content of 2.0 wt%. The support was slurried with water, and an appropriate amount of K2PdCl4 was added to this suspension. Afterwards, pH was adjusted and maintained at 11 by addition of solid KOH. The suspension was refluxed for 1 h, after which the mixture was cooled, filtered, washed, dried and reduced with hydrogen flow (3.6 L h−1) at 300 °C for 3 h, and finally passivated in 1 % O2/N2.
Bimetallic 2.0 wt%Pd-x wt%Re catalysts supported on TiO2 were prepared by two different methods using ammonium perrhenate (NH4ReO4): (i) impregnation of the monometallic Pd catalyst with a Re precursor aqueous solution (SI) or (ii) catalytic reduction (CR), as described in [29]. In the SI method, the parent Pd/TiO2 catalyst was added to distilled water and a predetermined amount of NH4ReO4 was introduced in this suspension which was maintained under stirring at room temperature for 5 h. Then, the solution was evaporated and the catalyst precursor was dried in vacuum at 50 °C during 20 h, before reduction at 450 °C for 3 h and passivation under 1 % O2/N2 flow. The second approach involved surface redox reaction (CR) occurring between hydrogen activated on the parent Pd/TiO2 catalyst and ammonium perrhenate (NH4ReO4) solution according to the overall redox reaction: 7Pd–Hads + ReO4 − + H+ → Re0(Pd)7 + 4H2O. The parent Pd/TiO2 catalyst was placed in a fixed bed reactor, reactivated under H2 flow (3.6 L h−1) at 300 °C for 1 h and then cooled down to room temperature while maintaining the H2 flow. Subsequently, the rhenium solution (acidified with HCl, pH = 1), previously degassed under N2 bubbling, was introduced onto the catalyst at room temperature. After 1 h reaction time under H2 bubbling, the solution was filtered out, and the catalyst was dried overnight at 100 °C under H2 flow (3.6 L h−1). Finally, the bimetallic catalyst was reduced under hydrogen flow (3.6 L h−1) at 450 °C for 3 h before storage in ambient air.
The actual Pd and Re contents in the prepared catalysts were determined by ICP-OES (inductively coupled plasma optical emission spectrometer, Perkin) with an accuracy of ±0.1 wt%. The different metal loadings in the catalyst references given in the text are directly derived from the ICP analysis results. The metallic accessibility was determined by H2 chemisorption using a pulsed technique. The catalysts were reduced in H2 flow (1.8 L h−1) at 300 °C for 1 h, then flushed by Ar flow (1.8 L h−1) at the same temperature for 2 h, and finally cooled to 70 °C before H2 pulses. To avoid formation of β-Pd hydride phase, chemisorption of H2 was performed at 70 °C. Powder X-ray diffraction (XRD) patterns of the catalysts were measured for 2θ comprised between 10° and 80° (step of 0.03°, step time of 2 s) using a Bruker AXS D5005 X-ray diffractometer and a CuKα radiation (λ = 1.54184 Å) as X-ray source. Phase identification was made by comparison with JCPDS database.
2.2 Hydrogenation Reaction
All experiments were performed in a Hastelloy Parr 4560 high pressure reactor of 300 mL equipped with an electrically heated jacket, a turbine agitator with a magnetic driver, and a liquid sample line. In a typical reaction, the reactor was loaded with 100 mL of a 5 wt% SUC aqueous solution (420 mmol L−1) and 1 g catalyst (molar ratio SUC/Pd = 225). After purging with Ar, the reactor was heated to 160 °C. The aqueous samples taken from the reactor at regular intervals were analyzed using both gas chromatography (HP-5 column, 30 m × 0.25 mm column, thickness 0.25 μm) and a high performance liquid chromatography instrument equipped with UV and RI detection (ICSep Coregel 107H column at 40 °C, 0.005 N H2SO4 as mobile phase at a flow rate of 0.5 mL min−1). The main reaction products consisted of GBL, THF, and BDO. By-products analyzed in the liquid phase were n-butanol, n-propanol, butyric acid, and propionic acid (Scheme 1). The mass balance was checked by measuring total organic carbon (TOC) in the liquid phase using a Shimadzu TOC-VCSH analyzer. This measure indicates if significant C–C cracking reactions occurred, transferring compounds to the gas phase. Indeed, the difference in the TOC concentration introduced into the reactor and the measured TOC in the product solutions was an estimation of gaseous products formed. Some experiments were performed twice; the reaction rates and selectivity to the various products were reproducible.
3 Results and Discussion
No X-ray diffraction pattern was observed for 2.1 wt% Pd/TiO2 A and 2.1 wt% Pd/TiO2 B monometallic catalysts except that of TiO2 supports, suggesting the presence of very small Pd crystallites. However, the catalysts presented different metallic dispersions as measured by H2 chemisorption (28 and 16 %, respectively, using TiO2 A and TiO2 B as support).
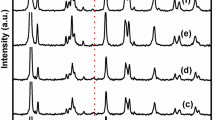
Using both methods of preparation for the bimetallic catalysts, the percentage of rhenium deposited was always 80–95 % of the introduced amount. The XRD patterns of bimetallic catalysts prepared by method SI are shown in Fig. 1. No diffraction peak of Pd species was observed. This was due to the good dispersion of Pd species. On the other hand, crystallites of potassium perrhenate KReO4 salt were detected in 2.2 wt%Pd–3.4 wt%Re/TiO2 A/SI (Fig. 1a). The presence of this phase could be attributed to the preparation method of the parent monometallic catalyst. In spite of thorough washing of the catalyst after preparation, potassium ions may remain on the catalyst and may be easily exchanged with ammonium ions. A bimetallic catalyst 2.2 wt%Pd–2.6 wt%Re/TiO2 A/SI was prepared on the monometallic catalyst after additional washing of the Pd monometallic catalyst. As seen in Fig. 1b the presence of KReO4 was no more detected.

Diffractogram of (a) 2.2 wt%Pd–3.4 wt%Re/TiO2 A/SI, (b) 2.2 wt%Pd–2.6 wt%Re/TiO2 A/SI, and (c) TiO2 A support. The diffraction lines correspond to the most intense ones of Pd. (asterisk) diffraction peaks assigned to KReO4 (JCPDS 004-007-5493)
XRD of the bimetallic catalysts prepared by CR indicated no characteristic peaks of Pd, as was observed in the monometallic parent catalysts. The CR deposition method conducted in aqueous acidic solution does not modify the dispersion of the monometallic Pd catalysts. This was further verified in blank experiments, in which the palladium monometallic catalysts were subjected to the conditions under which rhenium was added, except no rhenium salt was added. The final monometallic catalysts showed similar dispersions from H2 chemisorption (30 and 18 % on TiO2 A and TiO2 B, respectively). No salt of ReO -4 or any other Re species was detected in these catalysts which had been reduced at 450 °C and then handled in air. This observation suggests that the Re species are highly dispersed or in too low amounts to be detected. It is also in agreement with earlier published literature which concluded that Re mostly exists as highly dispersed clusters (<1 nm) in supported bimetallic catalysts [30, 31].
The behaviour of the catalysts was evaluated in the hydrogenation reaction of SUC. Figure 2a shows the evolution of the concentrations of compounds in the liquid phase as a function of time in the presence of the 2.1 wt% Pd/TiO2 A monometallic catalyst.

Hydrogenation of succinic acid in the presence of 2.1 wt% Pd/TiO2 A. a Evolution of main products as a function of time: filled circle SUC, filled square GBL, open triangle BDO, open diamond THF. b TOC concentration: open circle calculated from HPLC results, open square measured using TOC-analyzer
In the presence 2.1 wt% Pd/TiO2 A, SUC was progressively converted to attain 94 % conversion after 48 h. GBL was the main product detected over the monometallic catalyst. The type of TiO2 support (A or B) was found to play a critical role on the metallic dispersion and therefore on the catalytic activity. Indeed, 2.1 wt% Pd/TiO2 B behaved similarly, but it was much less active; conversion was only 75 % after 48 h. In the presence of both monometallic catalysts very low concentrations of BDO and THF were formed (<8 mmol L−1), and GBL was formed with selectivity >95 %. Negligible amounts of n-butanol were formed (<0.7 mmol L−1), whereas neither propionic acid nor butyric acid could be detected. Figure 2b shows the evolution of the TOC value as calculated from the HPLC analysis in the liquid phase and from direct analysis using the TOC-analyser. The measured TOC remained at a value around the initial TOC value introduced into the reactor. This shows that insignificant cracking reactions occurred. This observation is consistent with the negligible or nil measured concentrations of butanol, propanol, and butyric or propionic acids. In addition, the measured and calculated TOC values fit well within experimental uncertainty, which ascertains that all intermediates and final products in the liquid phase were identified and quantified. These observations are consistent with literature describing that palladium-based catalysts are efficient catalysts for selective formation of GBL from SUC either in dioxane [15, 16] or in water [13] solvent.
The hydrogenation was then performed on bimetallic catalysts prepared by depositing NH4ReO4 precursor on the monometallic catalysts, either by the SI or the CR method. A typical reaction profile of SUC hydrogenation using a bimetallic catalyst is shown in Fig. 3. The deposition of rhenium on the Pd/TiO2 A catalyst (by the CR method in that case) enhanced very significantly the activity of the catalyst. It should be noted that no reaction was observed when only Re deposited on TiO2 was present. SUC was completely converted within 24 h and during this step GBL was also the main product formed. Before total conversion of SUC, at roughly 10 h of reaction, GBL was at its maximum concentration at 260 mmol L−1, and BDO and THF (at a lesser extent) are produced at higher concentrations than in the presence of the monometallic catalyst. After total SUC conversion, GBL was converted further and yielded essentially BDO. After 48 h, the degree of conversion of GBL was 90 % and the selectivity to BDO and THF was 66 and 6.5 %, respectively. In addition, n-butanol, n-propanol, butyric, and propionic acids were formed as further by-products. However, they were all present in very low concentrations in the liquid phase.

Hydrogenation of SUC in the presence of 2.2 wt%Pd–0.8 wt%Re/TiO2 A/CR. a Evolution of main products concentrations as a function of time: filled circle SUC, filled square GBL, open triangle BDO, open diamond THF. b Evolution of by-products concentrations: filled diamond n-butanol, filled triangle n-butyric acid, plus n-propanol, times n-propionic acid. c TOC concentration: open circle calculated from HPLC results, open square measured using TOC-analyzer
It is worth noting that the measured carbon content remained approximately constant at the introduced value (ca. 420 mmol L−1 C4 equivalent), which reveals that no significant cracking reactions occurred. Also, the carbon contents calculated from HPLC and GC analysis were close to the experimental values, demonstrating again that all products in liquid phase were analyzed.
The bimetallic catalysts with different percentages of Re and prepared by both methods were then compared on both supports. Figure 4 shows the results for the bimetallic catalysts with various Re loadings using both methods of deposition. The loadings of 3.4 and 3.6 wt% Re for the SI-bimetallic catalysts were chosen according to the previous results on Pd–Re catalysts prepared on active carbon supports [13]. Also further, comparison of 2.2 wt%Pd–3.4 wt%Re/TiO2 A/SI (Fig. 4) and 2.2 wt%Pd–2.6 wt%Re/TiO2 A/SI (not shown) revealed that the latter was less active.

Comparison in succinic acid hydrogenation of bimetallic catalysts prepared on: (a–d) TiO2 A: open diamond monometallic catalyst, filled triangle 2.2 wt%Pd–1.9 wt%Re/RC, filled circle 2.2 wt%Pd–0.8 wt%Re/CR, and filled square 2.2 wt%Pd–3.4 wt%Re/SI; (e–h) TiO2 B: open diamond monometallic catalyst, filled triangle 2.0 wt%Pd–1.5 wt%Re/RC, filled circle 2.0 wt%Pd–0.6 wt%Re/CR, and filled square 2.0 wt%Pd–3.6 wt%Re/SI
Compared to the monometallic catalysts, the addition by the SI method of 3.4 or 3.6 wt% Re on Pd/TiO2 and Pd/TiO2 B, respectively, greatly enhanced the reaction rate of SUC transformation. On TiO2 A, conversion was complete within 10 h. The rate was lower on support TiO2 B, because of the lower dispersion of the parent Pd catalyst. The presence of the rhenium in the solid led to a drastic change in the product distribution. Over the bimetallic catalysts, GBL was easily hydrogenated leading to BDO and THF with selectivity greatly in favor of BDO. After 48 h, GBL conversion was nearly complete on TiO2 A with a final selectivity to BDO as high as 83 %. On TiO2 B, the GBL hydrogenation rate was lower. In that case, after 48 h, GBL concentration was still >50 mmol L−1, but selectivity to BDO was close to the one measured on TiO2 A. The effect of increasing the Re loading using the CR method is also shown in Fig. 4. Differently from what was observed over the SI prepared bimetallic catalysts, the addition of only a small concentration of Re additive was sufficient to achieve a synergy between Pd and Re. In spite of the low Re loadings, the TiO2 A and TiO2 B supported Pd–Re catalysts containing 0.8 and 0.6 wt% Re, respectively, exhibited reasonably higher reaction rates for SUC conversion than the monometallic catalysts. However, this acceleration was lower than in the case of the respective SI catalysts tested. Furthermore, these catalysts showed lower efficiency for further GBL hydrogenation. They also seem to be slightly less selective for BDO, and a slightly greater amount of THF was systematically formed for a given remaining GBL concentration to be hydrogenated. As a consequence, the selectivity to BDO is expected to be a few percentages lower on the CR prepared catalysts. The catalytic activity of CR catalysts was much dependent on the amounts of Re loaded. With too much Re addition, i.e. Re loadings of 1.9 and 1.5 wt% on TiO2 A and TiO2 B, respectively, the catalysts showed much lower activity and their behavior was closer to that of monometallic catalysts. It was concluded that a low amount of Re deposited using the CR method was very efficient in modifying the monometallic catalysts to be selective to BDO. However, the SI method exhibited the best performances. These results provide clear evidence that the Re presence is essential for high BDO production and that the activity is strongly related to the addition mode of the rhenium salt and the amount of Re deposited onto the catalyst. It was also observed a very good mass balance for all experiments (not shown). These results suggest that no excessive C–C cracking reactions to gaseous products occurred.
Characterization of Pd–Re catalysts is not an easy task. It has been shown in previous literature that the covering of Pd metallic particles with rhenium species, the interactions between the two metals, and the extent of palladium and rhenium depend on the support, the metal precursors, the reduction temperatures [30–33]. Usually, the rhenium species are not fully reduced to metallic state. Indeed, the reducibility of oxidic rhenium on different supports has been much discussed, but no consensus has been reached yet [31, 33–37]. The catalysts used in this work are presently characterized using transmission electron microscopy (TEM) with energy dispersive X-ray spectroscopy (EDS), temperature-programmed reduction (TPR), XPS, Raman to address these questions, in particular to explain the different behavior of both types of preparations (SI or CR). It is beyond the scope of this paper to discuss them in detail and further information is necessary. The results will be the published in a subsequent paper. Nevertheless, some preliminary observations may be mentioned. Quantitative analysis of the H2 consumption during chemisorption experiments suggest that contact during catalyst preparation was established. The results are similar to the findings for Pd–Re/Al2O3 system [38]. EDS scans of particles show evidence of the presence of both metals in particles, though some particles appear to contain only Pd or Re. It is also generally accepted that a mixture of Re species with different oxidation states are present after a reduction treatment under H2 flow [31]. Using XPS spectroscopy, we noted that some re-oxidation of the metals occurred if the catalysts were exposed to air and handled in air after reduction at 450 °C under H2. However, during the liquid phase hydrogenation reaction under pressure, some in situ-reduction of the catalysts must occur, which may still change the fraction of each Re species [33].
Finally, from the above results, it can be seen that the hydrogenation reaction from SUC to BDO is a two step-reaction via the GBL. The second step contributes to the diol formation. In order to confirm the above results on the different capacities of the catalysts to form BDO, some of the catalysts were evaluated in the direct hydrogenation of the substrate GBL under the same conditions (400 mmol L−1, 160 °C, 150 bar, 1 g catalyst, 120 g solution). Figure 5 compares the catalysts prepared on both supports for GBL disappearance and for BDO formation. The THF concentration was always less than 40 mmol L−1 after 48 h (not represented). The results confirm the higher activity for GBL hydrogenation of catalysts prepared on both supports by the SI method. It is also clear that catalysts prepared from TiO2 A exhibited higher activity in this reaction.

Hydrogenation of γ-butyrolactone (GBL, plain symbols) to 1,4-butanediol (BDO, empty symbols) in the presence of : (filled diamond, open diamond) 2.2 wt%Pd–3.4 wt%Re/TiO2 A/SI (filled square, open square) 2.2 wt%Pd–0.8 wt%Re/TiO2 A/CR, (filled triangle, open triangle) 2.0 wt%Pd–3.6 wt%Re/TiO2 B/IS, and (filled circle, open circle) 2.0 wt%Pd–0.6 wt%Re/TiO2 B/CR
4 Conclusions
The results obtained over mono and bi-metallic catalysts showed that rhenium addition to 2.0 wt% Pd/TiO2 catalysts is essential for selective hydrogenation of SUC to BDO. A synergy exists between Pd and the Re species which simultaneously enhances the activity for the SUC and intermediate GBL hydrogenation reactions to yield BDO. The Re loading to reach maximum BDO production changed according to the bimetallic catalyst preparation used. A high loading (>3.5 wt% Re) was necessary using the successive impregnation method. The minimum Re loading requirement for modification of redox bimetallic catalysts was found much lower (0.6–0.8 wt% Re). Selectivity to BDO higher than 83 % was accomplished at total conversion of SUC and GBL.
References
Werpy T, Petersen G (eds) (2004) Top value-added chemicals from biomass: volume 1-results of screening for potential candidates from sugars and synthesis gas. Department of Energy, Washington
Bechthold I, Bretz K, Kabasci S, Kopitzky R, Springer A (2008) Chem Eng Technol 31:647
Werpy T, Frye J, Holladay J (2008) In: Kamm B, Gruber PR, Kamm M (eds) Biorefineries-industrial processes and products: status quo and future directions vol 2. Wiley-VCH Verlag GmbH, Weinheim, p 367
Bozell JJ, Petersen GR (2010) Green Chem 12:539
Turner K, Sharif M, Rathmell C, Kippax JW, Carter AB, Scarlett J, Reason AJ, Harris N (1988) US 4751334 to Davy MacKee London
Felthouse TR, Burnett JC, Mitchell BF, Mummey MJ (2001) Maleic anhydride, maleic acid, and fumaric acid. In: Kirk-Othmer encyclopedia of chemical technology. John Wiley and Sons Inc., New York, p 893
Varadarajan S, Miller DJ (1999) Biotechnol Prog 15:845
Cukalovic A, Stevens CV (2008) Biofuels Bioprod Biorefin 2:505
Delhomme C, Weuster-Botz D, Kühn FE (2009) Green Chem 11:13
Toba M, Tanaka S-I, Niwa S-I, Mizukami F, Koppany Z, Guczi L, Cheah K-Y (1999) Appl Catal A Gen 189:243
Deshpande RM, Buwa VV, Rode CV, Chaudhari RV, Mills PL (2002) Catal Commun 3:269
Luque R, Lin CSK, Du C, Macquarrie DJ, Koutinas A, Wang R, Webb C, Clark JH (2009) Green Chem 11:193
Pham Minh D, Besson M, Pinel C, Fuertes P, Petitjean C (2010) Top Catal 53:1270
Tachibana Y, Masuda T, Funabashi M, Kunioka M (2010) Biomacromolecules 11:2760
Hong UG, Lee J, Hwang S, Song IK (2011) Catal Lett 141:332
Hong UG, Hwang S, Seo JG, Lee J, Song IK (2011) J Ind Eng Chem 17:316
Hong UG, Park HW, Lee J, Hwang S, Yi J, Song IK (2012) Appl Catal A 415–416:141
Song H, Lee SY (2006) Enzyme Microbial Technol 39:352
McKinlay JB, Vieille C, Zeikus G (2007) Appl Microbial Biotechnol 76:727
Orjuela A, Yanez AJ, Peereboom L, Lira CT, Miller DJ (2011) Sep Purif Technol 83:31
Beauprez JJ, De Mey M, Soetaert WK (2010) Process Biochem 45:1103
Chaudhari RV, Rode CV, Deshpande RM, Jaganathan R, Leib TM, Mills PL (2003) Chem Eng Sci 58:627–632
WO 2011/123269 and WO 2011/123270 to BioAmber S.A.S
Some patents for hydrogenation in aqueous solution: US 4550185 (1985), US4609636 (1986), US4782197 (1988), US 5478952 (1995), US 6008384 (1999), and US 6670490 (2003) to E.I. Du Pont de Nemours; US 5473086 (1995), US 5698749 (1997), and US 5969164 (1999) to Standard Oil Co, US 6204417 (2001) to BASF, US 6989455 (2006) to ISP Investments
Rylander PN (2001) In: Hydrogenation methods (best synthetic methods). Academic Press, London, p 78
Pintar A, Besson M, Gallezot P (2001) Appl Catal B 31:275
Zhang Z, Jackson JE, Miller DJ (2008) Bioresour Technol 99:5873
Epron F, Especel C, Lafaye G, Marecot P (2008) In: Astruc D (ed) Nanoparticles and catalysis. Wiley-VCH Verlag GmbH&Co, Weinheim, p 279
Vuurman MA, Stufkens DJ, Oskam A (1992) J Mol Catal 76:263
Bare SR, Kelly SD, Vila FD, Boldingh E, Karaopetrova E, Kao J, Mickelson GE, Modica FS, Yang N, Rehr JJ (2011) J Phys Chem C 115:5740
Meitzner G, Via GH, Lytle FW, Sinfelt JH (1987) J Chem Phys 87:6354
Manyar HG, Paun C, Pilus R, Rooney DW, Thompson JM, Hardacre C (2010) Chem Commun 46:6279
Johnson MFL, LeRoy VM (1974) J Catal 35:434
Yao HC, Shelef M (1975) J Catal 44:392
Wang L, Hall WK (1983) J Catal 82:177
Mitra B, Gao XT, Wachs IE, Hirt AM, Deo G (2001) Phys Chem Chem Phys 3:1144
Ziemecki SB, Jones GA, Michel JB (1986) J Catal 99:207
Acknowledgments
This work was supported by the French Agence Nationale de la Recherche within the Programme Chimie et Procédés pour le Développement Durable CP2D 2009 (HCHAIB).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ly, B.K., Minh, D.P., Pinel, C. et al. Effect of Addition Mode of Re in Bimetallic Pd–Re/TiO2 Catalysts Upon the Selective Aqueous-Phase Hydrogenation of Succinic Acid to 1,4-Butanediol. Top Catal 55, 466–473 (2012). https://doi.org/10.1007/s11244-012-9813-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-012-9813-3