
Abstract
Chromium(III), zinc(II) and nickel(II) complexes of thio-Schiff base derived from salicylaldehyde and 4-amino-2,4-dihydro-1,2,4-triazole-5-thione have been encapsulated in the nanopores of zeolite-Y by a flexible ligand method. The prepared encapsulated metal complexes have been characterized by surface analysis (XRD and N2 adsorption/desorption), spectroscopic methods, chemical and thermal analyses. The various techniques of characterization used demonstrated that these complexes were effectively encapsulated in the zeolite supercages without structural modification or loss of crystallinity of the zeolite framework. The encapsulated complexes were screened as heterogeneous catalysts for various oxidation reactions such as phenol, cyclohexene and styrene oxidation, using H2O2 as an oxidant. Under the optimized conditions, these catalysts exhibited high to moderate activity. After a few cycles these catalysts were found to be stable and could be reused after recovering without detectable catalyst leaching or significant loss of activity.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Heterogeneous catalysts provide new routes for environmentally benign processes. In this respect, encapsulation/immobilization of metal complexes in porous materials, especially zeolites, gained much interest in the last few decades [1–7]. The encapsulation complexes serve as functional mimic of enzymes, possessing many advantageous features of both homogeneous and heterogeneous catalysis.
Upon physical entrapment of metal complexes in the supercages of zeolites, it is termed ship-in-a-bottle complexation. It is observed that they cannot only retain their high activity and selectivity in homogeneous catalysis but also exhibit high stability. This is due to the reduction of degradation of the organic ligands and dimerization of the transition metal complexes in cavities [8–13], which could occur during homogeneous catalytic reactions, resulting in a reduction in the activity, and even irreversible deactivation.
Numerous studies have been conducted on encapsulation of a variety of metal complexes [14, 15], clusters [16, 17] and organometallic compounds [9, 18] in zeolitic hosts. Intensive studies have been considered to improve the catalytic properties for these materials in the oxidation of different types of organic substrates using alternative oxidation processes based on green and non-toxic oxidants. This is one of the main challenges to overcome by chemical industry from an economical and environmental viewpoint.
Thus far, considerable development has been achieved and many types of ligands (salen types, macrocyclic, etc.) have been used to prepare different types of such materials [18–32]. Many of these materials exhibit high activities in some industrially important reactions and possess the advantages of heterogeneous catalysis, such as easy separation and handling, efficient recycling, minimization of metal leaching and in some cases higher selectivity than homogeneous catalysts. We recently reported on the synthesis of thio-Schiff base ligand, abbreviated as [sal(thiotriazol)], which was used for encapsulation with different metal ions in zeolite-Y [33]. Herein, we report on another aspect of our investigation involving the synthesis of new encapsulated metal thio-Schiff base complexes of Cr(III), Ni(II) and Zn(II) in zeolite-Y (Structure 1) and screened their catalytic activity for various oxidation reactions, such as oxidation of phenol, cyclohexene and styrene.

Thio-Schiff base complexes
2 Experimental
2.1 Materials
All the reagents were of analytical grades (Sigma-Aldrich) and used without further purifications. High purity grade of solvents were dried and stored over molecular sieves. Zn(II), Ni(II) and Cr(III) nitrates, phenol, styrene, cyclohexene (Sigma-Aldrich) were distilled and dried. Salicylaldehyde, chromium(III), nickel(II) and zinc(II) nitrate, were purchased from Aldrich. Zeolite Na-Y was purchased from Fluka.
2.2 Physical Measurements and Analysis
The ATR-IR measurements were carried out on a Perkin-Elmer Spectrum 100 FTIR spectrometer. Electronic spectra were recorded on a GBC UV/VIS 920 UV–Visible spectrophotometer in absolute ethanol or in Nujol (by layering the mull of the sample to the inside of one of the cuvette while keeping another one layered with Nujol as reference). The elemental analyses were performed at University of Cape Town, South Africa. The percentage metal contents of the encapsulated complexes were determined by Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES) at the University of Stellenbosch, Cape Town, South Africa. 1H NMR spectra were recorded in CDCl3 using a Varian XR200 spectrometer. Sample signals are relative to the resonance of residual protons on carbons in the solvent. The nitrogen adsorption/desorption and BET surface area was determined at −196 °C using a Tristar 3000 Micromeritics. All samples were degassed prior to the measurement at 120 °C for 12 h. Powder X-ray diffraction (XRD) was recorded by Bruker AXS D8 Advance, high-resolution diffractometer with Cu Kα radiation (λ = 1.5406 Å) with PSD Vantec gas detector, at iThemba Labs, Cape Town, South Africa.
All catalyzed reaction products were analyzed using Agilent 7890 gas chromatograph fitted with flame ionization detector. A HP-5 (phenylmethylsilicon) capillary column (30 m × 330 pm × 0.25 μm film thickness, Agilent technologies). The retention time of all peaks was compared with authentic samples and also the identity of the products was further confirmed by a GC–MS using Finnigan MAT GCQ GC/Mass spectrometer.
2.3 Preparations of the Encapsulated Complexes
The encapsulated metal complexes were prepared using a two steps method (i) the preparation of metal ion exchange zeolite and (ii) the insertion of ligand in the cavity of the zeolite, followed by complexation with metal ions.
2.3.1 Preparation of the Ligand
The thio-Schiff base ligand [sal(thiotriazol)] was synthesized according to our reported procedure [33].
2.3.2 Preparation of Metal Exchanged Zeolite, M-Y
The ion-exchanged Na-Y zeolites were prepared using ion-exchange procedure by exchanging Na ions of Na-Y (5.0 g suspended in 300 mL deionized water) with 50 mmol aqueous nitrate solutions of the corresponding metal cations (Cr(III), Zn(II) or Ni(II)). The mixture was stirred at 90 °C for 24 h, filtered, washed with copious amount of hot deionised water followed by Soxhlet extraction with acetonitrile for 1 h till the filtrate was free from any metal ion content. The resulting precipitate was dried at 150 °C in air for 24 h.
2.3.3 Preparation of Zeolite-Y Encapsulated Metal Complexes
An amount of 1.0 g M-Y and 2.5 g of the ligand [sal(thiotriazol)] were mixed in a 50 mL MeCN following the general flexible ligand method [19, 20]. The suspension mixture was refluxed overnight. The obtained slurry was extracted by Soxhlet in acetonitrile for 48 h then treated with 0.01 M NaCl solution. The encapsulated complexes were filtered, washed with copious amount of hot distilled water and dried at 150 °C for several hours.
2.4 Catalytic Studies
2.4.1 Hydroxylation of Phenol
Oxidation of phenol using the prepared catalysts was carried out in 50 mL glass parallel reactor vessels (Radley’s Discovery Technologies 12 place Heated Carousel Reaction Station fitted with a Reflux unit). In a typical reaction, phenol (4.7 g, 0.05 mol) and 30% solution of H2O2 (5.67 g, 0.05 mol) were mixed in 2 mL of the desired solvent and the reaction mixture was heated at 80 °C with continuous stirring. Toluene was added as internal standard. An appropriate amount of catalyst (0.010 g) was added to the reaction mixture, the reaction was considered to begin and this was assumed as the starting point of the reaction. The progress of the reaction was monitored by GC analysis using an internal standard technique. The reaction products were analyzed using a gas chromatograph and monitored at set time intervals by withdrawing small aliquots. Samples were filtered before analysis. The identities of the products were confirmed by GC–MS.
2.4.2 Oxidation of Cyclohexene
Cyclohexene (0.82 g, 10 mmol), aqueous 30% H2O2 (2.27 g, 20 mmol) were taken in acetonitrile (2 mL). The reaction mixture was allowed to heat at 80 °C for 6 h with continuous stirring. The catalyst (0.010 g) was added to the reaction mixture and the reaction was considered to begin. The analysis and identification of the reaction products were done as mentioned above.
2.4.3 Oxidation of Styrene
In a typical oxidation reaction, styrene (0.52 g, 5 mmol) and aqueous 30% H2O2 (1.13 g, 5 mmol) were mixed in 2 mL of acetonitrile and the reaction mixture was heated at 80 °C in an oil bath. The catalyst (0.010 g) was added to the reaction mixture, and this was assumed as the starting point of the reaction. The analysis and identification of the reaction products were done as mentioned above.
3 Results and Discussion
3.1 Syntheses and Characterisation of Catalysts
Synthesis of Cu(II), Ni(II) and Zn(II) thio-Schiff base complexes, encapsulated in the nanocavity of zeolite-Y, was carried out by the flexible ligand method. The ligand diffused through the zeolite supercages, followed by complexation with metal ions. Complexation of the ligand with Cr(III) and Ni(II) ion-exchanged zeolite-Y was accompanied by a colour change (green and pale green, respectively), while the encapsulated Zn(II) complex was colourless.
Soxhlet extraction in acetonitrile was used to remove remaining uncomplexed ligand in the cavities as well as that located on the external surface of the zeolite, in addition to the free complex. The remaining uncomplexed metal ions in zeolite were removed by exchanging back with aqueous 0.01 M NaCl solution. The product was washed with copious amounts of hot distilled water until the filtrate was free from chlorides. Thus, the metal content estimated by ICP is exclusively due to encapsulation of the complex in the supercages of the zeolite-Y.
The synthesized catalysts were characterized by FTIR and electronic spectra, thermal analysis and XRD, which supported the encapsulation of metal complexes inside the cavities of the zeolite-Y.
The parent NaY zeolite possesses a Si/Al molar ratio of 3, which corresponds to a unit cell formula Na48(SiO2)144(AlO2)48·xH2O. The encapsulated complexes exhibited a similar Si/Al ratio to the parent Na-Y zeolite, which indicates negligible change in the zeolite framework after encapsulation. The encapsulated complexes were well purified using Soxhlet extraction, and as a result the metal and element contents found after encapsulation are only due to the presence of the metal complexes in the cavity of the zeolite-Y.
The metal complex per unit cell was calculated from the estimated metal content in the encapsulated complex and corresponds to 4.7 for Ni-based, 7.6 for Cr-based and 1.3 for Zn-based catalysts, where x = ~250 water molecules.
Elemental and chemical analysis data confirmed the stoichiometry of the encapsulated metal complexes (Table 1). The chemical analyses of the samples revealed the presence of an organic moiety with a C/N ratio. The M/N ratio suggested a 2:1 ligand-to-metal stoichiometry analogous to the model complexes with formula [CrL2(H2O)Cl], [NiL2·2H2O], and [ZnL2].
The IR spectra of the metal-encapsulated complexes are presented in Fig. 1. The intensities of IR bands of the encapsulated complexes are weak due to low concentrations of the complex encapsulated in zeolite cavities. The absorption peaks of the encapsulated complexes can only be detected in the region where the zeolite Na-Y bands typically do not absorb, specifically in the regions 1600–1200 and 350–450 cm−1.

FTIR spectral pattern of zeolite Na-Y and the encapsulated catalysts
IR spectra of NaY zeolite, metal-exchanged zeolites presented strong zeolite lattice bands around 1140, 1035, 960, 780 and 740 cm−1. No significant broadening or shift of the structure-sensitive zeolite vibrations at 1140 cm−1 (attributed to the asymmetric stretching vibrations of (Si/Al)O4 units) and other zeolite bands, after ion-exchange and upon encapsulation of the complexes, were observed. This implies that the zeolite framework remained unchanged upon the encapsulation of the complexes, indicating that there is no defect of zeolite framework or dealumination, therefore the matrix remained unchanged during the process, in agreement with XRD results.
The IR spectrum of the ligand [sal(thiotriazol)] showed characteristic bands at 3132, 2715 and at ~1110 cm−1, which are assigned to ν(N–H), ν(S–H) and ν(C=S), respectively [34]. These observations suggest that the Schiff base exhibited thiol-thione tautomerism, which is well-known within all of the other members of this family of triazoles [35–38].
The phenolic OH vibration of the ligand (3332 cm−1) disappeared in that of the complexes, indicating the involvement of this group in coordination with the metal ion, via deprotonation. A strong band at 1623 cm−1 in the spectrum of the ligand assigned to ν(C=N) was lowered by (10–15) cm−1 upon complexation. This is due to the coordination of the azomethine nitrogen with the metal ion.
A broad band in the region 3400–3520 cm−1, and two weaker bands in the region 750–800 and 700–720 cm−1, in all complexes indicated the presence of coordinated water [34]. New moderately intense bands appearing in the low frequency region (350–500 cm−1) in all encapsulated complexes originate from (M–O) and (M–N) stretching vibrations. This further indicates the coordination of nitrogen and oxygen to the metal. The bands due to C=S and S–H vibrations of the ligand remained unperturbed in the encapsulated complexes. These observations suggest the non-involvement of sulphur atoms in coordination [39].
On the basis of FTIR data, it s concluded that all metal ions are coordinated to the azomethine nitrogen, phenolic oxygen, and water molecules or/and chloride. Thus, IR data indicates the encapsulation of the complexes in the zeolite cavity.
The encapsulation of metal complexes inside the cavity of zeolite-Y is further confirmed by UV–Vis spectroscopy, which was recorded over the range 200–900 nm as shown in Fig. 2.

Electronic spectra and UV–Vis spectral data of the encapsulated complexes
All encapsulated complexes exhibit bands between 380 and 410 nm due to symmetry forbidden metal-to-ligand charge-transfer (MLCT) band. Other bands appearing at 280–325, 250–265, 202–220 nm for all complexes arise due to intra-ligand n–π*, π–π* and ϕ–ϕ* transitions, respectively.
The pale green coloured [CrL2(H2O)Cl]-Y exhibits a weak and broad asymmetric band centred at 580 nm, ascribed to a d–d transition due to 4A2g(F) → 4T2g(F) (ν 1). The band at 380 nm is assigned to 4A2g(F) → 4T1g(F) (ν 2) transitions in an octahedral fields. The band due to 4A2g(F) → 4T2g(P) transition appears at 270 nm. All bands compare well with the reported value for [Cr(H2O)6]3+ [40].
The absorption spectrum of [NiL2·2H2O]-Y exhibits a band at 314 nm, which is clearly due to 3A2g → 3T1g(P) (ν 3) transition as a majority of complexes exhibit a ν 3 band in this region [40]. As expected, the zinc complex exhibits only intra-ligand bands. On the basis of spectral evidences the square planar structure for the catalysts encapsulated in the zeolite-Y has been suggested.
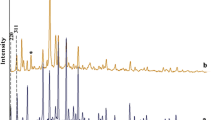
XRD patterns of the encapsulated complexes were recorded to study their crystallinity and to ensure encapsulation. The encapsulated complexes exhibit similar diffraction peaks to those of M-NaY and parent Na-Y zeolite; except for a slight change in the intensity of the peaks, no new crystalline pattern emerges. These observations again confirm that the framework and crystallinity of zeolite-Y have not undergone any significant structural changes during the encapsulation, and that the complexes fit well into the cages. The relative peak intensities of the 220, 311 and 331 reflections have been considered to be related to the locations of cations which indicated that significant cation redistribution occurred followed by complex formation within the zeolite supercages [41].
It is clear from the XRD pattern that the line intensities of the I 331 > I 220 > I 311 for the Na-Y zeolite, but I 331 > I 311 > I 220 for the encapsulated complexes. Only one new peak in Ni and Zn-based catalysts is observed with a d-value of 1.764 nm (at 2θ = 52.5°) and 16.902 nm (at 2θ = 17.8°), respectively. This is also located in the corresponding metal ion-exchanged-Y at the same position.
Two new peaks with d-values of 3.392 nm (at 2θ = 29.2°) and 2.181 nm (at 2θ = 46.0°) were observed in Cr-Y as well as in [CrL2]-Y. This could be attributed to atomic level integration resulting in non-vanishing structure factor for certain peaks, which were extinct in Na-Y structure. This clearly shows the importance of the extraction during the encapsulation process to ensure the removal of the surface species and non-complexed metal ions. This information clearly indicates the insertion of metal complexes in the cavities of the zeolite.
Figure 3 shows the N2 adsorption/desorption isotherms of the encapsulated catalysts, which are typical type I according to the IUPAC classification [37], and are characteristics of the microporous nature of the materials. The surface area, pore volume and pore size of the encapsulated metal complex, along with that of the parent Na-Y zeolite, are presented in Table 2. A noticeable reduction in the surface area, pore volume and adsorption capacity of the zeolite was observed upon encapsulation of metal complexes in the supercages of zeolite-Y. This supports the observation that the complexes are present within the zeolite cages and not on the external surface since the zeolite crystallinity was retained. However, the decreasing values in the surface area, pore volume and adsorption capacity depends on the amount of incorporated complexes as well as their molecular size and geometrical conformation inside the zeolitic host.

N2 adsorption/desorption isotherms of the encapsulated catalysts
The thermogravimetric analyses data of the encapsulated metal complexes, along with the percentage mass loss at different steps and their probable assignments, are listed in Table 3 and presented in Fig. 4. Clearly, all the encapsulated complexes exhibit three weight loss regions. Both Cr- and Zn-based catalysts exhibit similar first and second steps of weight loss: 25–130 °C (trapped water) and 130–210 °C (intra-zeolite water). Though all catalysts were dried at 150 °C, it is expected that even at this temperature intra-zeolite water remains in these zeolites. The third step between 210 and 750 °C for the Cr-based catalyst consists of overlapping steps assigned to the loss of the coordinated chloride ion, a water molecule and the chelating ligand, while the Zn-based catalyst shows single step loss in this region, which is assigned to the ligand decomposition.

TGA profile of the encapsulated complexes
The Ni-based catalyst also shows three step loss at 25–230, 230–330 and 330–610 °C due to trapped and intra-zeolite water molecules, two coordinated water molecules and the slow decomposition of the chelating ligand, respectively.
A very small percentage of weight loss indicates the presence of only a small amount of metal complex inserted in the zeolite matrix. This is in agreement with the low-percentage metal content, as estimated by ICP.
3.2 Catalytic Performance of the Encapsulated Metal Complexes/Zeolite-Y
The synthesized catalysts were screened for various oxidation reactions under optimized reaction conditions.
3.2.1 Hydroxylation of Phenol
Figure 5 shows the liquid-phase hydroxylation of phenol using the synthesized encapsulated catalysts and H2O2 as an oxidant as a function of time under the optimized reaction conditions, which we reported recently (i.e. 0.05 mol phenol, 1:1 phenol/H2O2 molar ratio, 10 mg catalyst, 2 mL CH3CN at 80 °C) [33]. Two products, catechol (CAT) and hydroquinone (HQ), were observed with a mass balance of >95%. Other products could not be detected by the gas chromatograph. The phenol conversion using these catalysts enhances with the reaction time and attains a steady state after 5 h, giving maximum phenol conversion (30–33%). The oxidation proceeded rapidly during the first 1 h and reached a maximum of 20–26% conversion. Though the reaction is fast in the first hour; all these catalysts perform much better in the next 2 h. In terms of selectivity these catalysts show a CAT/HQ ratio of 3 after the first hour of the reaction time, and decreases to half its value upon increasing reaction time to 12 h. Thereafter, only a very slow conversion was observed up to 24 h.

% Conversion/selectivity for hydroxylation of phenol using various catalysts
Comparing the catalytic activities of these catalysts with other metal-encapsulated complexes in zeolite-Y analogues, our catalysts produced higher conversion than Zn, Ni and Cr(salpn)-Y [31], and comparable to [26, 28] and other encapsulated Zn complexes [41, 42].
No significant changes in the activity and product selectivity was observed after acquiring a steady state ~12 h. Very small amounts (<2%) of two additional products were also observed with all the catalysts in the reaction mixture, but no attempts were made to identify them.
3.2.2 Oxidation of Cyclohexene
The catalytic results for the oxidation of cyclohexene over the synthesized catalysts, in the presence of H2O2, are shown in Fig. 6. With regards to product distribution, the reaction solution gave 2-cyclohexen-l-ol, 2-cyclohexen-1-one, cyclohexeneoxide and 1,2-cyclohexediol as a major products (Scheme 1). The product distribution changed with time and was different for the various catalysts used in this study. All the catalysts show a considerable selectivity towards CHO. It seems that the zeolite not only protects the complex but also increases the epoxide yield.

Percentage conversion/selectivity for oxidation of cyclohexene over various catalysts

Oxidation of cyclohexene over encapsulated metal complexes
Both Ni and Cr-based catalysts show high activity in comparison to Zn-based catalysts. This decrease in activity may be due to the clogging of the pores of the matrix as a result of the trapping of the major CHdiOL reaction product (larger in size than others) within the zeolite, as reported by Herron [42, 43].
The Ni-based catalyst shows an excellent selectivity towards CHONE over the reaction period. While the Cr-based catalyst shows a different trend in selectivity towards the products. After the first 5 h of reaction, CHOL and CHO were the major products with 63–52 and 20–37% selectivity, respectively. After 12 h reaction time the selectivity towards CHO increased to 48%. The formation of the allylic oxidation products (i.e. 2-cyclohexene-1-one and 2-clohexene-1-ol) shows preferential attack of the activated C–H bond over the C=C bond.
No significant change in the activity and product selectivity is observed after acquiring a steady state ~12 h. Very small amounts (<2%) of two additional products were also observed with all the catalysts in the reaction mixture, but no attempts were made to identify them.
It can be concluded that the catalytic oxidation ability of these catalysts solely depends on the central metal ion present in the encapsulated complex, and not on the H2O2 decomposition ability.
Comparing the conversion of these catalysts with Ni-macrocyclic complexes encapsulated in zeolite-Y, our catalysts exhibited significantly higher activity [43, 44].
3.2.3 Oxidation of Styrene
Oxidation of styrene over the synthesized catalysts with H2O2 as an oxidant gave styreneoxide; benzaldehyde; 1-phenylethane-1,2-diol; and phenylacetaldehyde (Scheme 2).

Oxidation of styrene over encapsulated metal complexes
It is interesting to note that all these catalysts show high activity for oxidation of styrene with high selectivity towards 1-phenylethane-1,2-diol (40–65%) after 12 h reaction time (Table 4). The high yield of 1-phenylethane-1,2-diol is possibly due to hydrolysis of styreneoxide and to the oxidation of styreneoxide by a nucleophilic attack of hydroperoxy species on styreneoxide, followed by cleavage of the intermediate hydroperoxistyrene. Benzaldehyde may be produced via radical oxidative cleavage of the styrene side chain double bond. Other products such as phenylacetaldehyde may form through further oxidation of benzaldehyde or through isomerisation of styreneoxide.
The catalytic activity or conversion increases with reaction time and reached 45–63% after 1 h. Significant increases in the performance of these catalysts were observed on increasing the reaction time and level-off after 5 h (~90%). Further increase in reaction time up to 12 h enhanced the catalysts’ performance to 94–98%.
Table 4 provides the selectivity of various products using H2O2 as oxidant. In the case of [CrL2(H2O)Cl]-Y, the selectivity of various reaction products follows the order: 1-phenylethane-1,2-diol > benzaldehyde > phenylacetaldehyde > styreneoxide, while for [NiL2(H2O)2]-Y catalyst the order is: 1-phenylethane-1,2-diol > styreneoxide > benzaldehyde = phenylacetaldehyde; [ZnL2]-Y, the order is 1-phenylethane-1,2-diol > styreneoxide > benzaldehyde > phenylacetaldehyde.
Comparing the activity for the oxidation of styrene our catalysts shows much higher activity than the encapsulated macrocyclic compounds reported [45].
3.3 Reusability of the Catalyst
The recyclability of the encapsulated catalysts was tested for the oxidation of phenol cyclohexene and styrene. At the end of the reaction, the zeolite catalyst was recovered by filtration, regenerated by Soxhlet extraction (MeCN) for 1 h, dried at 120 °C overnight, and reused twice under similar reaction conditions. The recovered catalyst shows almost similar activity and selectivity after 12 h reaction time. For example, the recovered Ni-catalyst showed 32.9 and 32.6% phenol conversion after the first and second cycle, respectively. Whereas, the recovered Cr-catalyst displayed 81.4 and 80.3% cyclohexene conversion. On the other hand, recovered Zn-catalyst exhibited 97.8 and 97.7% styrene conversion. These results verify that the catalysts are highly stable and can be recycled. Moreover, the ICP analyses for the reaction filtrate show the absence of metal ions which indicate that no leaching has occurred during the reaction. This proved that the reaction is catalyzed heterogeneously.
4 Conclusion
The thio-Schiff base complexes of Cr(III), Ni(II) and Zn(II) have been successfully incorporated into the supercages of zeolite (α-cages), as evidenced by various physico-chemical studies.
The heterogeneous catalysts described in this paper are environmentally benign in liquid phase oxidations of phenol, cyclohexene and styrene using H2O2 as a clean oxidant.
These catalysts show excellent conversion for the oxidation of cyclohexene and styrene and moderate conversion towards hydroxylation of phenol under optimized reaction conditions. All these catalysts show high selectivity towards catechol formation (~70%) in the hydroxylation of phenol. In cyclohexene oxidation, the selectivity towards various products varied relative to the type of the encapsulated transition metal. The Ni-based catalyst shows high selectivity towards CHONE (~75%), while Cr- and Zn-based catalysts show high selectivity towards the other products (CHO, CHOL and CHdiOL). For oxidation of styrene, these catalysts were highly selective towards the formation of 1-phenylethane-1,2-diol.
The recycling studies demonstrate that these catalysts behave truly as heterogeneous catalysts.
In summary, this study proves that these encapsulated thio-Schiff base complexes are promising catalysts for various oxidation reactions.
References
Čejka J, Corma A, Zones S (eds) (2010) Zeolites and catalysis: synthesis, reactions and applications. Wiley-VCH, Weinheim
De Vos DE, Dams M, Sels BF, Jacobs PA (2002) Chem Rev 102:3615
Song CE, Lee S-G (2002) Chem Rev 102:3495
Sheldon RA, Arends IWCE, Lampers HEB (1998) Catal Today 41:387
Sheldon RA, van Bekkum H (2001) In: Fine chemicals through heterogeneous catalysis. Wiley-VCH, New York
Parton R, De Vos D, Jacobs PA (1992) In: Derouane EG, Lemos F, Naccache C, Riberio FR (eds) Proceedings of the NATO advanced study institute on zeolite microporous solids: synthesis, structure and reactivity. Kluwer Academic, Dordrecht, 555 pp
Martin-Aranda RM, Čejka J (2010) Top Catal 53:141
Medina JC, Gabriunas N, Paez-Mozo N (1997) J Mol Catal A 115:233
Kowalak S, Weiss RC, Balkus KJ Jr (1991) J Chem Soc Chem Commun 57
Herron N, Tolman CA (1987) J Am Chem Soc 109:2837
Pires EL, Magalhaes JC, Schuchardt U (2000) Appl Catal A 203:231
Jacob CR, Varkey SP, Ratnasamy P (1998) Microporous Mesoporous Mater 22:465
Kimura T, Fukuoka A, Ichikawa M (1990) Catal Lett 4:279
Srinivas D, Sivasanker S (2003) Catal Surv Jpn 7:121
Thomas JM, Raja R (2005) Ann Rev Mater Res 35:315
Chavan SA, Srinivas D, Ratnasamy P (2002) J Catal 212:39
Yahiro H, Kyakuno T, Okada G (2002) Top Catal 19:193
Gellin P, Naccache C, Taarit YB (1988) Pure Appl Chem 8:1315
Knops-Gerrits P, De Vos D, Thibault-Starzyk F, Jacobs PA (1994) Nature 369:543
Bowers C, Dutta PK (1990) J Catal 122:271
Balkus KJ Jr, Khanmamedova AK, Dixon KM, Bedioui F (1996) Appl Catal A 143:159
Raja R, Ratnasamy P (1997) J Catal 170:244
Ernst S, Selle M (1999) Microporous Mesoporous Mater 27:355
Grommen R, Manikandan P, Gao Y, Shane T, Shane JJ, Schoonheydt RA, Weckhuysen BM, Goldfarb D (2000) J Am Chem Soc 122:11488
Mesu JG, Visser T, Beale AM, Soulimani F, Weckhuysen BM (2006) Chem Eur J 12:7167
Maurya MR, Titinchi SJJ, Chand S (2004) J Mol Catal A 214:257
Maurya MR, Titinchi SJJ, Chand S (2003) J Mol Catal A 201:119
Maurya MR, Titinchi SJJ, Chand S (2003) Catal Lett 89:219
Maurya MR, Titinchi SJJ, Chand S (2003) J Mol Catal A 193:165
Maurya MR, Titinchi SJJ, Chand S (2002) Appl Catal A 228:177
Maurya MR, Titinchi SJJ, Chand S, Mishra IM (2002) J Mol Catal A 180:201
Maurya MR, Kumar M, Titinchi SJJ, Abbo HS, Chand S (2003) Catal Lett 86:97
Titinchi SJJ, Abbo HS (2010) Top Catal 53:254
Sen AK, Singh G, Singh K, Handa RN, Dubey SN, Squattrito PJ (1998) Proc Indian Acad Sci Chem Sci 110:75
McCarrick M, Eltzroth MJ, Squattrito PJ (2000) Inorg Chim Acta 311:95
Escobar-Valderrama JL, Garcia-Tapia JH, Ramirez-Ortiz J, Rosales MJ, Toscano RA, Valdes-Martinez J (1989) Can J Chem 67:198
Kajdan TW, Squattrito PJ, Dubey SN (2000) Inorg Chim Acta 300–302:1082
Weng NS (1992) Acta Crystallogr C48:2224
Geary WJ (1971) Coord Chem Rev 7:81
Cotton FA, Wilkinson G (1993) In: Advance Inorganic Chemistry, 3rd edn. Wiley Eastern, New Delhi, p 838
Derouane EG, Haber J, Lemos F, Guisnet M, Ribeiro FR (1998) In: Catalytic activation and functionalisation of light alkanes: advances and challenges. Kluwer Academic, Dordrecht, The Netherlands
Puttappa B, Kadidal N, Mahendra N (2010) J Porous Mater 17:107
Herron N (1989) New J Chem 13:761
Salavati-Niasari M (2008) Polyhedron 27:3132
Bansal VK, Thankachan PP, Prasad P (2010) Appl Catal A 381:8
Acknowledgment
The authors are thankful to the Research Committee of the University of the Western Cape for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abbo, H.S., Titinchi, S.J.J. Metallo Salicylidenetriazol Complexes Encapsulated in Zeolite-Y: Synthesis, Physicochemical Properties and Catalytic Studies. Top Catal 53, 1401–1410 (2010). https://doi.org/10.1007/s11244-010-9600-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-010-9600-y