
Abstract
Zeolite encapsulated amino acid complexes such as Fe-proline, Fe-histidine and Cu-valine have shown significant promise in the selective oxidation of hydrocarbons such as cyclohexane and benzylic alcohols. This novel encapsulation procedure, which involves building a zeolite (host) around a pre-formed and structurally defined amino acid complex (guest), results in the generation of isolated single-sites for catalytic oxidations. IR and UV–Vis techniques were used for structure determination as well as establishing the integrity of the synthesised complexes. Preliminary results indicate that complexes were formed by coordination to the nitrogen atom of the amino group and the oxygen atom of the carboxylate group to the metal. The hydrophobic/hydrophilic nature of the zeolite, coupled with the high activity of the neat complex, generates highly active and selective single-site heterogeneous catalysts for the production of cyclohexanone, an important precursor to the production of commodity chemicals such as adipic acid and ε-caprolactam.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
There has been a growing interest in the study of interactions of transition metal cations with biomolecules, such as amino acids, primarily because of their propensity to catalyze the functionalization of C–H and C–C bonds. Complexes of transition metals and amino acid residues are at the core of numerous biological processes, such as dioxygen transport, electron transfer, and oxidation or hydrogenation of target compounds [1]. In these processes, the active site of the enzymes are highly specific and often involve one or several histidine residues interacting with transition metal cations.
Monooxygenases are the enzymes capable of selectively catalyzing oxidation reactions, where one oxygen atom is added to a hydrocarbon molecule, usually producing a hydroxyl group, and the other atom is used to form water. Generation of this very active oxygen species is the most remarkable feature of monooxygenases. Mononuclear iron active sites catalyze a wide variety of reactions, including the hydroxylation of aliphatic C–H bonds, the epoxidation of C–C double bonds, the cis-dihydroxylation of arene double bonds and heterocyclic ring formation [2]. The mononuclear iron site usually consists of three ligands from protein residues, two histidines and one carboxylate, around the iron centre as shown in Scheme 1 [3]. Binuclear iron enzymes (see Scheme 2) involved in O2 activation primarily exist in two oxidation states: the fully reduced bi-ferrous [FeII]2 and the oxidized bi-ferric [FeIII]2 form. A particular example of a di-iron containing enzyme is methane monooxygenase (MMO) [4], in which the binuclear iron site is capable of producing oxygen species of much superior activity than other monooxygenases. In addition to aromatics and other compounds, MMO exhibits a unique ability to convert even methane, which is known to be a very inert organic molecule, into methanol using dioxygen as the oxidant.

Structure of the mono-iron active site of monooxygenase. The iron atom is coordinated by two histidine (His), bidentately by aspartic acid (Asp) and water molecule

Coordination geometries for the diiron centre of MMOHred from M. capsulatus
The immobilization of metal complexes within solid matrices has proven to be an important step in heterogeneous enzymatic catalysis. Mono [5] and binuclear [6] iron complexes supported on solid supports have been used with dioxygen in the selective oxidation of hydrocarbons with many types of ligands, such as porphyrin [7], salen (N,N′-bis(salicylidene)ethylenediamine) [8], phthalocyanines [9], tris-(pyridylmethyl) amine(TPA) [10] and (bis-(2-pyridylmethyl)amine (BMPA) [11] being used to synthesize different types of such catalysts (Fig. 1). These materials exhibit high activity, favourable selectivity, and are considered to be good functional models for metalloenzymes.

Functional models of iron centres for hydrocarbon oxidation
1.1 Metal Amino Acid Complexes
Amino acids as protein constituents are small molecules with various functional groups. They are good metal-complexing agents, forming chelates through the amino and carboxylate groups. In addition, amino acids often have a side chain with a metal binding group, such as the imidazole group of histidine, the phenol ring of tyrosine, and the carboxylate group of aspartate and glutamate, which serve as the metal binding sites in most proteins. At high pH, the carboxyl group tends to be dissociated, giving the compound a stronger negative charge while at low pH, the amino group is protonated shifting the molecule to a net positive charge. At the isoelectric point, the amino acid in solution, has a net charge of zero. At this interim pH, a solution contains positively and negatively charged amino acid ions in equal quantities called zwitterions (Scheme 3).

Dissociation equilibrium in amino acids
1.1.1 l-histidine
l-histidine ((S)-2-amino-3-(4-imidazolyl) propionic acid) is a very strong metal-coordinating ligand (see Fig. 2a) and plays an important role in the binding of the metal ions in proteins [12]. l-histidine contains three potential coordination sites, namely the carboxylate oxygen (Ocarboxyl), the imidazole nitrogen (Nim), and the amino nitrogen (Nam). The imidazole nitrogen of the aromatic histidine provides the primary metal binding site in proteins and is a versatile molecule in enzyme catalysis. At physiological pH values (7–8), the imidazole group can readily switch between different states, protonated or non-protonated, and catalyse the formation, or the breaking, of chemical bonds. Histidine can occur in several pH dependent states of protonation and coordination, either as a part of the peptide backbone, or as a free ligand molecule. When the pH value is increased, the amine proton is deprotonated to give a mononegative anionic ligand. On the other hand, when the pH value is lowered, the Nim and Ocarboxyl are respectively protonated. As a result of this, l-histidine can act as a mono-, bi-, or tridentate ligand and its mode of coordination implicitly depends on the pH of the solution. At low pH, l-histidine forms a mono complex with the metal species and at high pH, a bis complex is obtained. With metal ions having a square-planar coordination sphere, for steric reasons, only two of the three binding sites can coordinate resulting in either histamine-like or glycine-like complexes (Fig. 2b).

Structure of l-histidine, a potential tridentate ligand (a) and structures of metal–histidine complexes which bear resemblance to histamine and glycine (b)
1.1.2 l-proline
l-proline ((S)-Pyrrolidine-2-carboxylic acid) is unique among the amino acids because the α-amino group (NH) is secondary—see Fig. 3a. The side chain is bonded to the nitrogen of the α-amino group, as well as the α-carbon atom. Proline is sometimes called an imino (NH) acid rather than an amino (NH2) acid because of the bonding to the nitrogen atom and is also a neutral and non-polar amino acid. The N–H group starts to deprotonate around pH 8, and forms a strong complex with the metal at pH 10 (Fig. 3b). l-proline can occur in several pH dependent states of protonation—at low pH, l-proline exists in a cationic form, in high pH in an anionic form and as a zwitterion at the isoelectric point (Fig. 3b). Because proline lacks hydrogens on the amide group, it cannot act as a hydrogen bond donor, but only as a hydrogen bond acceptor. The imino (NH) group is fixed rigidly in the pyrrolidine ring limiting the mobility of the N–H bond with respect to the carboxyl group. This rigidity is one of the factors that drives the important role played by proline in the folding of proteins, where steric affects arising from the proline side chains help to determine the stabilities and positions of the protein folds [13].

Structure of bis-proline (a) and depiction of the various pH dependant states of protonation (b)
A number of heterogeneous proline-based catalysts have been reported.
l-Proline and its chiral complexes with zinc have shown to act as enantioselective catalysts for aldol reactions, selenenylation of aldehydes, and the synthesis of bromoesters and benzimidazoles [14, 15].
1.1.3 l-valine
l-valine (l-2-Amino-3-methylbutanoic acid) is a nonpolar amino acid with an aliphatic side chain. l-valine is hydrophobic and is usually found in the interior of the protein. At high pH, the carboxyl group of l-valine is dissociated, while at low pH, the amino group is protonated, and at the isoelectric point, l-valine in solution, has a net charge of zero (zwitterions) (Fig. 4b). l-valine coordinates to the metal ion through the amino and carboxylate groups forming a five-membered chelate ring as shown in Fig. 4a.

Structure of bis-valine (a) and depiction of the various pH dependant states of protonation (b)
In our search for novel and efficient oxidation catalysts, we found that natural amino acids, which are protein constituents, are effective biomimetic alternatives when used as ligands. Transition-metal-containing amino acid complexes can serve as isolated, active single-sites [16–18] when encapsulated in inorganic host materials such as zeolites. In this article, we will describe a novel, improved ‘build-bottle-around-ship’ trategy for encapsulating metal-containing amino acid complexes within the cavities of the zeolite X to exploit their catalytic potential in the oxidation of cyclohexane and benzyl alcohol.
2 Experimental
2.1 Synthesis of Proline Complex with Iron
Fe(proline)2 complex was prepared by the reaction of iron acetate (1 eq.) with proline (2 eq.) [19]. l-proline (0.49 g, 4.34 mmol) was dissolved in 10 mL of MeOH. To this solution 0.6 mL of triethylamine (Et3N) was added and the mixture was stirred for 0.5 h. The addition of iron acetate (0.37 g, 2.17 mmol) resulted in the formation of a yellow-grey precipitate. After stirring for 1 h the resulting precipitate was collected by filtration, washed with methanol and dried in the air (see Scheme 4).

Synthetic strategy for the preparation of Fe(proline)2 complex
2.2 Encapsulation of l-proline Complex with Iron in Zeolite X
The Fe(proline)2 complex was encapsulated within the supercages of Zeolite-X using the ‘zeolite synthesis method’ [20]. The silicate gel was prepared by stirring the 1.4 g of fumed silica, 1.06 g of NaOH, 0.1 g of Fe(Proline)2 and 3 mL of water. Addition of the aluminate solution (aluminium isopropoxide 3 g, NaOH 1.06 g and water 2 mL) resulted in a pale-orange slurry. The slurry was transferred to a polypropylene bottle with stirring for 24 h at room temperature and subsequently heated at 80 °C for 15 h. Complexes adsorbed on the exterior surface were removed by Soxhlet extraction technique. The resulting solid was dried at 80 °C for 24 h.
2.3 Synthesis of l-histidine Complex with Iron
l-histidine (1.55 g, 0.01 mol) was dissolved in 10 mL of 1N NaOH solution and slowly mixed with solution of FeSO4∙7H2O (1.39 g, 0.005 mol) in 20 mL of water. The brown-orange precipitate was almost instantaneous, but stirring was continued for 30 min. The complex was filtered, washed with 1:1 water:acetone mixture and dried in the air.
2.4 Encapsulation of l-histidine Complex with Iron in Zeolite
The Fe-histidine complex was encaged in zeolite Y using the ion-exchange method [21]. 0.6 g of Na-exchanged zeolite-Y was stirred for 6 h in a Fe(His)2 solution containing 3.192 × 10−4 M FeSO4∙7H2O and 1.6 × 10−3 M l-histidine. The pH of Fe(His)2 solution was adjusted to 7.3 by using NaOH or HCl solution. After ion exchange, the sample was separated from the solution by filtration, washed with water and dried at 60 °C overnight in the air.
2.5 Synthesis of l-valine Complexes with Copper
2.5.1 Cu(l-valine)2
A total of 1.17 g of l-valine (0.01 mol) was dissolved in 10 mL of 1N NaOH solution and slowly mixed with 25 mL of CuSO4 water solution (1.245 g, 0.005 mol). The blue crystalline precipitate was filtered, washed with 1:1 water:acetone mixture and dried in air.
2.6 Encapsulation of Copper l-valine Complex in Zeolite X
The Cu(valine)2 complex was encapsulated within the supercages of zeolite X using the zeolite synthesis method, as outlined earlier in Sect. 2.2.
2.7 Catalyst Characterisation
FTIR spectra were recorded using a Nicolet 380 spectrometer to confirm the structural integrity of the neat and encapsulated complexes. X-Ray powder diffraction patterns were collected employing a Siemens D5000 diffractometer using Cu Kα1 radiation. The UV–Vis spectra were recorded on Perkin–Elmer Lambda 900 DR UV–Vis with WinLab 900 software. N2 adsorption measurements were performed at liquid nitrogen temperature. Samples were first dehydrated and degassed in vacuum at 80 °C. Surface area and micropore volume were determined by nitrogen adsorption-desorption isoterms using Micromeritics ASAP 2020. The surface area was calculated using the Brunauer–Emmett–Teller (BET) method and the pore size was calculated using the Barrett–Joyner–Helenda (BJH) method.
2.8 Catalytic Oxidation
The aerobic oxidation of cyclohexane of the neat complexes and their corresponding zeolite-encapsulated heterogeneous analogues were performed in a high-pressure, stainless-steel catalytic reactor (Parr 4842) and comparative experiments with tert-butyl hydroxyperoxide (70 wt%) were carried out at atmospheric pressure using a glass-lined reactor. In both cases, the catalytic oxidations were studied at 98 °C. The reaction products were analysed by GC (Varian model 3400 CX) employing a HP INNOWAX capillary column and Flame Ionisation Detector (FID). The identity and the quantification of the products were established by using mesitylene as an internal standard using the calibration method.
3 Results and Discussion
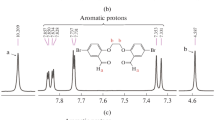
The structure of the proposed Fe(l-proline)2, Fe(l-histidine)2 and Cu(l-valine)2 amino acid complexes used in this study are shown in Fig. 5. The IR spectra of the neat metal complexes were analyzed in comparison to that of their corresponding free ligands and crosschecked with the literature data [22–24]. The IR spectra of the complexes show changes in the positions and the profiles of some bands, as compared to those of the free amino acid ligands (Table 1). The IR spectra of the neat Fe(l-proline)2 indicate that the complex was readily formed by coordination of the nitrogen atom of the amino group and the oxygen atom of the carboxylate group to the iron centre. The l-proline molecule is in its anionic form with the δNH2 + vibration missing when it is chelated to the metal, but vibrations associated with the COO− and the NH groups are present. The bands around 3,050 cm−1 identify the NH2 + symmetric stretching and 1,550 cm−1 band reflecting NH2 + scissoring confirm the protonation in the imino group of free l-proline. As was expected, these bands disappeared in the spectrum of the complex, and the NH stretching at 3,207 cm−1, suggests coordination through the NH group (indicate electron donation of the amine group to the iron). The neutral molecular form has vibrations specifically due to the stretches of the C=O group around 1,790 cm−1 and 1,766 cm−1 and does not exhibit vibrations due to the NH2 + or COO− groups. Because the COO− group is present in both the zwitterion form and in the complex, the C=O group virations are not observed. This result indicates that carboxylic group was not protonated in the complex and acts as metal binding site, else, a strong band at about 1,790 cm−1 should have been observed. The carboxylate stretching band at 1,607 cm−1 (νas COO−) was shifted to 1,660 cm−1, and the νs COO− bands at 1,448 cm−1 to 1,417 cm−.

Proposed structure of Cu(l-valine)2 (a); Fe(l-proline)2 (b) and possible models for l-histidine coordination in bis complexes with iron (c–f): c Nam(1)Nim(1)Nam(2)Nim(2), d Nam(1)Nim(1)Nim(2)OCOO(2), e Nam(1)Nim(1)Nam(2)OCOO(2), f Nam(1)OCOO(1)Nam(2)OCOO(2) coordination, respectively.(pink-Fe, green-O, blue-N, grey-C)
In the spectra of the Cu(l-val)2 complex, NH2 bands are present at 3,269 cm−1 and 3,158 cm−1 as antisymmetric and symmetric stretching modes, respectively. This indicates the coordination to the Cu through NH2 group. The carboxylate ion group absorbs strongly near 1,600 cm−1 and more weakly near 1,400 cm−1 because of antisymmetric and symmetric stretching modes. In the l-valine ligand, νas COO− and νs COO− are observed at 1,601 cm−1 and 1,451 cm−1. In the Cu(l-val)2 complex the peak at 1,613 cm−1 is assigned to COO− antisymmetric stretching and the peak at 1,463 cm−1 is assigned to COO− symmetric stretching. Because C=O group stretches were not present, this indicates that the carboxylic group was not protonated and COO− group coordinates to the metal in the complex. An exceptional difference between the IR spectra of free ligand to that of the metal complex was that, in the latter, NH2 and COO− bands are sharp and well resolved.
The zeolite encapsulated Fe(l-proline)2 catalyst showed well-resolved, sharp XRD peaks confirming the Faujasitic architecture. X-Ray powder diffraction patterns (Fig. 6) of the neat zeolite (without complex) and the zeolite encapsulated Fe(l-proline)2 complex are in the excellent agreement with no phase-impurities or defect structures. The crystallinity and structural integrity of the Faujasite zeolite remains unaltered after the encapsulation process. The absence of structural reflections due to the neat complex further reveals the presence of the Fe(l-proline)2 complex within supercages of Zeolite-X. This was also independently confirmed using other techniques such as DR UV–Vis, surface area, pore-volume, and adsorption techniques.

Powder XRD patterns of Zeolite X (a) and Zeolite X encapsulated Fe(l-proline)2 (b)
The formation and encapsulation of the transition-metal amino acid complexes inside Zeolite-X were also supported by UV–Vis spectroscopy. The comparison of the UV spectra for Fe-histidine before and after metal incorporation showed two new bands at 35,000 and 42,000 cm−1, which can be assigned to ligand to metal charge transfer transitions (LMCT) [25]. In the UV–Vis spectra of encapsulated material bands, due to LMCT, also appeared at 38,000 and 47,000 cm−1, which further indicates the presence of the complex within zeolite cages (Fig. 7). Interestingly, in the case of the Fe(proline)2 complex, there was a drastic decrease in the surface area and pore volume in the encapsulated analogues, which further confirms the encapsulation of the amino acid complex within the zeolite cages (Table 2).

UV–Vis spectra of (1) Fe-histidine complex and (2) zeolite encapsulated Fe-histidine
The catalytic activity of the neat and encapsulated Fe(proline)2 complex was evaluated in the oxidation of cyclohexane and the results are summarised in Fig. 8. The neat Fe(proline)2 complex displayed a remarkable change in product selectivity, simply by changing the nature of the oxidant. Interestingly, the zeolite-encapsulated analogues showed an overwhelming preference for the desired cyclohexanone and virtually no cyclohexanol was produced when O2 was used as the oxidant. Preliminary results for the oxidation of benzyl alcohol (Table 3) indicate that, the zeolite encapsulation methodology has been highly successful, leading to the generation of powerful functional mimics of enzymes that catalyze the production of valuable fine-chemical intermediates such as benzaldehyde with high yields and high selectivities using benign conditions. Further catalytic tests are currently underway to establish the nature of the active site, the hydrophobic/hydrophilic influence of the host (zeolite) in steering the product selectivity and the underlying mechanism.

Results for the catalytic oxidation of cyclohexane using both neat and zeolite encapsulated Fe(proline)2 complexes in the presence of air and tert-butyl hydroperoxide as oxidants. Reaction conditions: cyclohexane 2 g (0.02 mol), TBHP 2.57 g (0.02 mol), air 3.0 MPa, T = 371 K
4 Summary and Future Prospects
Cyclohexanone, which is an industrially important commodity chemical and a vital precursor in the manufacture of ε-caprolactam (for nylon-6) and adipic acid (for nylon 6,6), can be produced in high selectivities using benign oxidants and novel zeolite-encapsulated amino acid complexes. These benign and green catalysts have shown tremendous potential in the oxidation of benzylic alcohols with high selectivities towards the corresponding aldehydes. The presence of well-defined and isolated single-sites, coupled with the hydrophobicity/hydrophilicity of the host, results in the generation of a biomimetic system that is powerful functional mimic of monooxygenase enzymes.
References
Knops-Gerritis PPHJM, Goddard WA III (2003) Catal Today 81:263
Chen K, Costas M, Que L Jr (2002) J Chem Soc, Dalton Trans 672
Que L Jr, Ho RYN (1996) Chem Rev 96:2607
Wallar BJ, Lipscomb JD (1996) Chem Rev 96:2625
Holland AW, Li G, Shahin AM, Long GJ, Bell AT, Tilley TD (2005) J Catal 235:150
Trukhan VM, Gritsenko ON, Nordlander E, Shteinman AA (2000) J Inorg Biochem 79:41
Costa AA, Ghesti GF, de Macedo JL, Braga VS, Santos MM, Dias JA, Dias SCL (2008) J Mol Catal A: Chem 282:149
Corrêa RJ, Salomão GC, Olsen MHN, Cardozo Filho L, Drago V, Fernandes C, Antunes OAC (2008) Appl Catal A: Gen 336:35
Sorokin AB, Mangematin S, Pergrale C (2002) J Mol Catal A: Chem 182:267
Kim J, Harrison RG, Kim C, Que L Jr (1996) J Am Chem Soc 118:4373
Carvalho NMF, Horn A Jr, Antunes OAC (2006) Appl Catal A: Gen 305:140
Deschamps P, Kulkarni PP, Gautam-Basak M, Sarkar B (2005) Coord Chem Rev 249:895
Mateo E, Brlow SM, Hag S, Raval R (2002) Surf Sci 501:191
Kehat T, Portnoy M (2007) Chem Commun 2823
Giacalone F, Gruttadauria M, Marculescu AM, Noto R (2007) Tetrahedron Lett 48:255
Thomas JM, Raja R, Lewis DW (2005) Angew Chem Int Ed 44:6456
Thomas JM, Raja R (2006) Topics Catal 40:3
Thomas JM, Raja R (2001) Chem Commun 675
Darbre T, Machuqueiro M (2003) Chem Commun 1090
Raja R, Ratnasamy P (1997) Catal Lett 48:1
Weckhuysen BM, Verberckmoes AA, Fu L, Schoonheydt RA (1996) J Phys Chem 100:9456
Moszczenski CW, Hooper RJ (1983) Inorg Chim Acta 70:71
Marti EM, Methivier Ch, Dubot P, Pradier CM (2003) J Phys Chem 107:10785
Herlinger AW, Long TV (1970) J Am Chem Soc 92:6474
Frunz L, Prins R, Pirngruber GD (2007) Chem Mater 19:4357
Acknowledgements
This research was supported by EPSRC (UK) and The British Italian Partnership Program. We also wish to thank David Xuereb for his valuable assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dzierzak, J., Lefenfeld, M. & Raja, R. Single-site Biomimetic Amino Acid Complexes for the Benign Oxidation of Hydrocarbons and Alcohols. Top Catal 52, 1669–1676 (2009). https://doi.org/10.1007/s11244-009-9305-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-009-9305-2